Flow Magnetic Tweezers Workflow - Addtional Information
Additional information
This page will explain in more detail how the features we are sorting by look like. To understand better how this sorting step works we will talk about it in more detail. So we bascially try to answer the question how does the program know that the molecule shown above is a singly-tethered, coilable and what force the molecule experiences? The threshold inputs which are used are shown here. They will be discussed in detail through rest of the page.
We will have a closer look on the trace which is displayed in the screenshot below. We will zoom in on all the important features of this particular trace. As long as a trace has similar features compared to this trace (within a certain range) it is consider for the final analysis. We will check out the features from the left to the right.
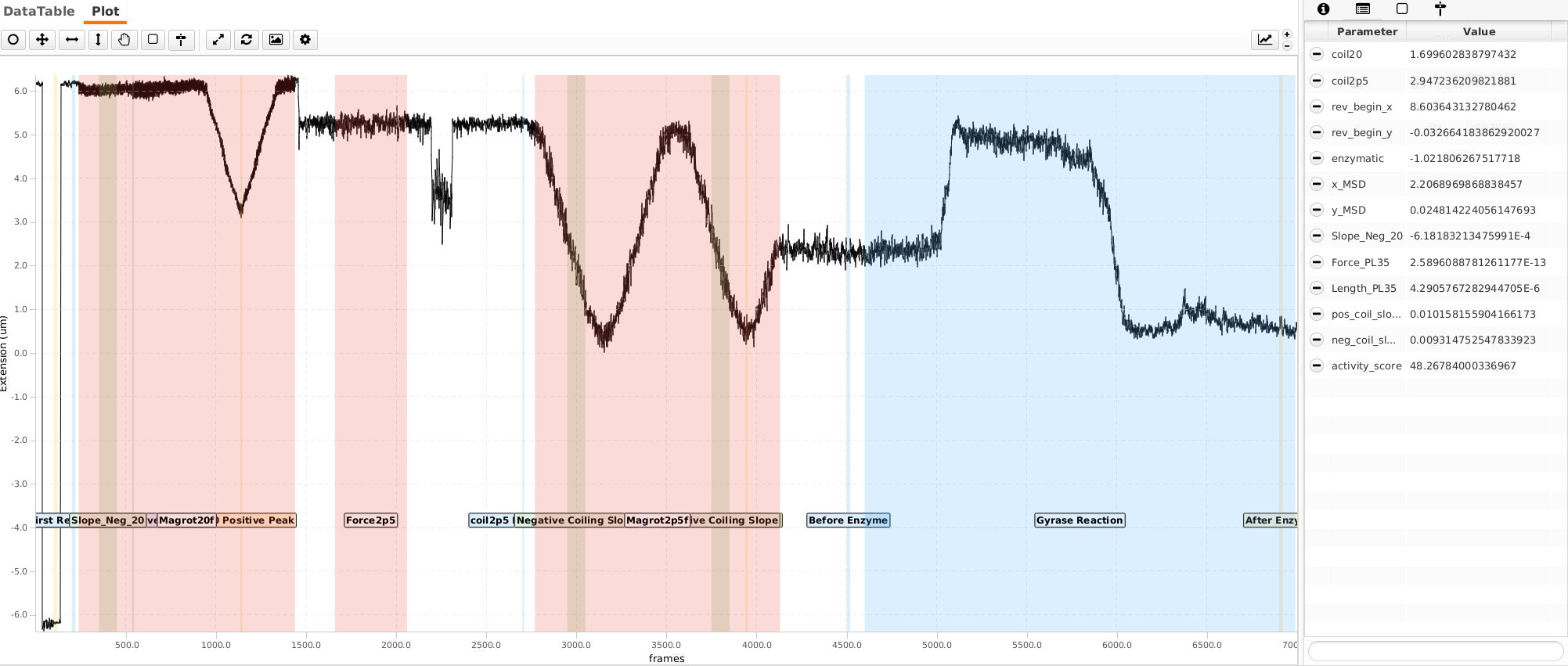
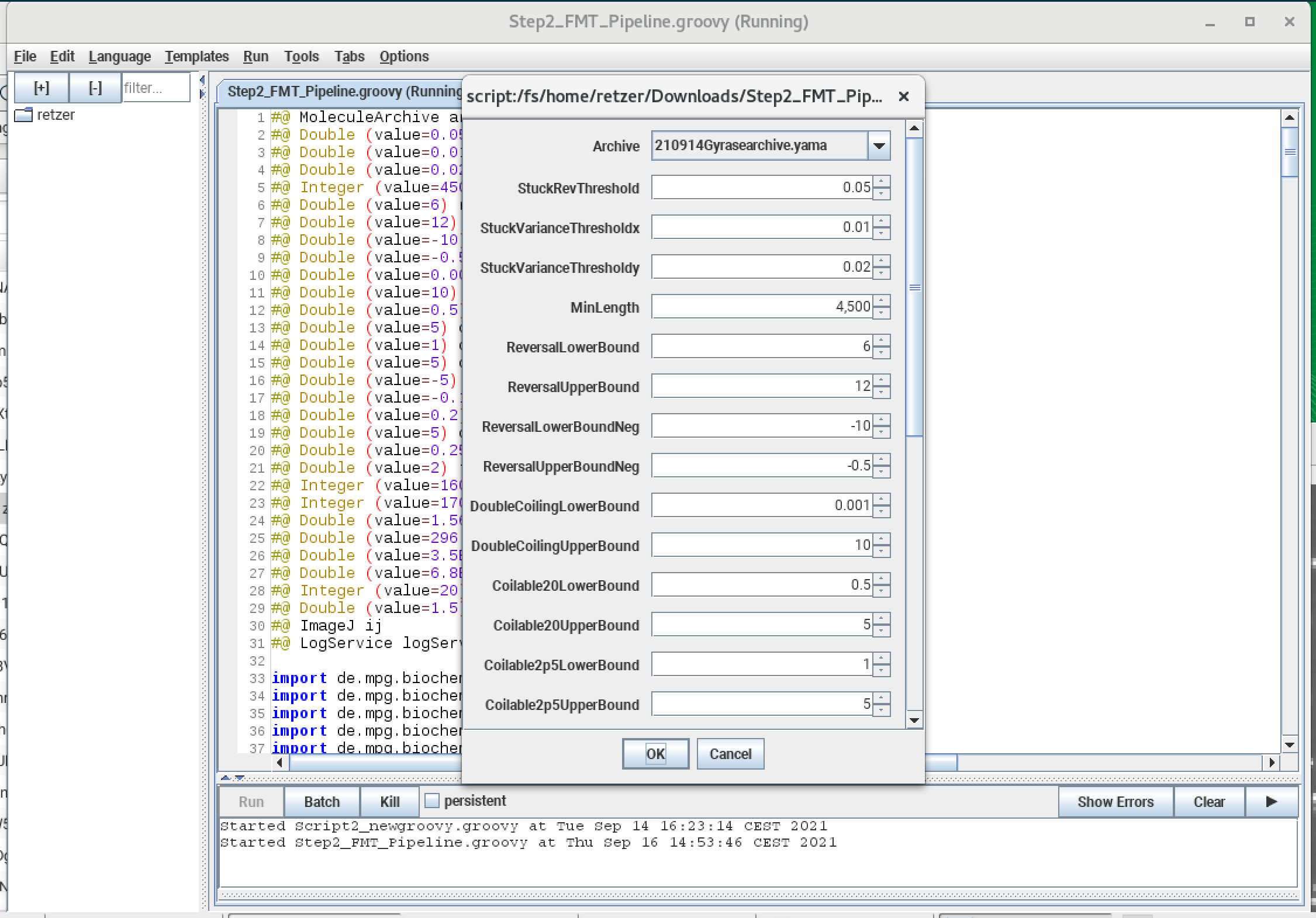
Values for this particular example are set by default in the script. They can be modified in the user interface which is also shown in the screenshot below.
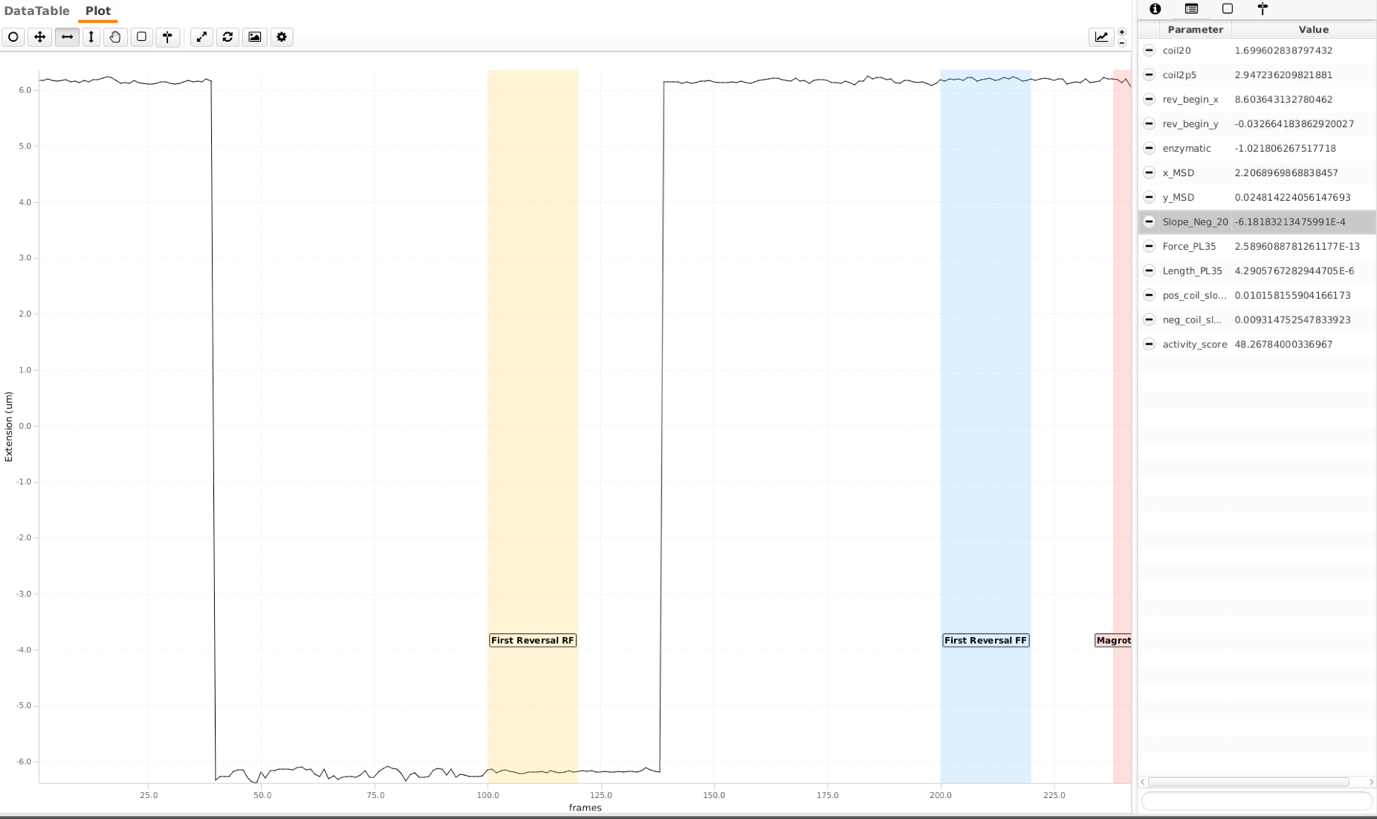
Reversal at the beginning
The zoom is set to initial part of the trace called reversal (starting from left side: beginning of the trace). The first 238 frames here show a flow reversal (the direction of the flow is reversed so the DNA tether will flip over). The regions are named first reversal RF (reverse flow) and first reversal FF (forward flow). An inbuilt function called ‘region difference calculator’ is used. As the name suggest, it calculates the difference between the stated region. Here the parameter was rev_begin_x (reversal at the beginning for x coordinate). It should be around twice the length of the DNA tether. The reversal threshold separate mobile beads from stuck beads. Also, some multiply-tethered beads can be classified using reversals.
//Get Regions form the metadata
First_Reversal_RF = metadata.getRegion("First Reversal RF")
First_Reversal_FF = metadata.getRegion("First Reversal FF")
//First Reversal: Adding the Parameters rev_begin_x and rev_begin_y
//RegionDifferenceCalculatorCommand.calcRegionDifference(molecule, xColumn, yColumn, regionOne, regionTwo, parameterName)
RegionDifferenceCalculatorCommand.calcRegionDifference(molecule, "slice", "x", First_Reversal_RF, First_Reversal_FF, "rev_begin_x")
RegionDifferenceCalculatorCommand.calcRegionDifference(molecule, "slice", "y", First_Reversal_RF, First_Reversal_FF, "rev_begin_y")
//reversal start
if (molecule.getParameter("rev_begin_x") > reversalLowerBound &&\
molecule.getParameter("rev_begin_x") < reversalUpperBound) {
molecule.addTag("mobile_begin")
}
//reversal start neg
if (molecule.getParameter("rev_begin_x") > reversalLowerBoundNeg &&\
molecule.getParameter("rev_begin_x") < reversalUpperBoundNeg) {
molecule.addTag("mobile_begin_neg")
}
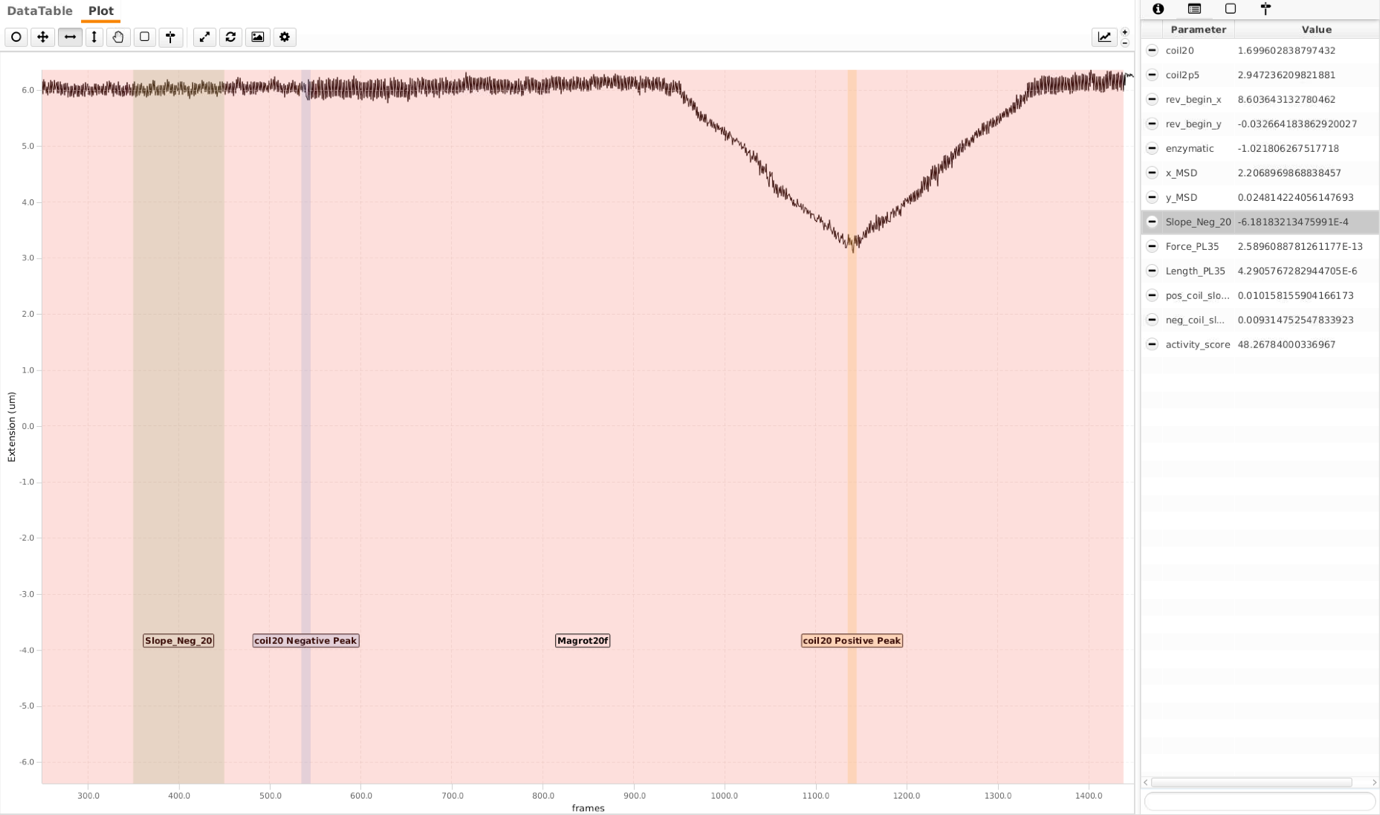
Magnet rotation at high force
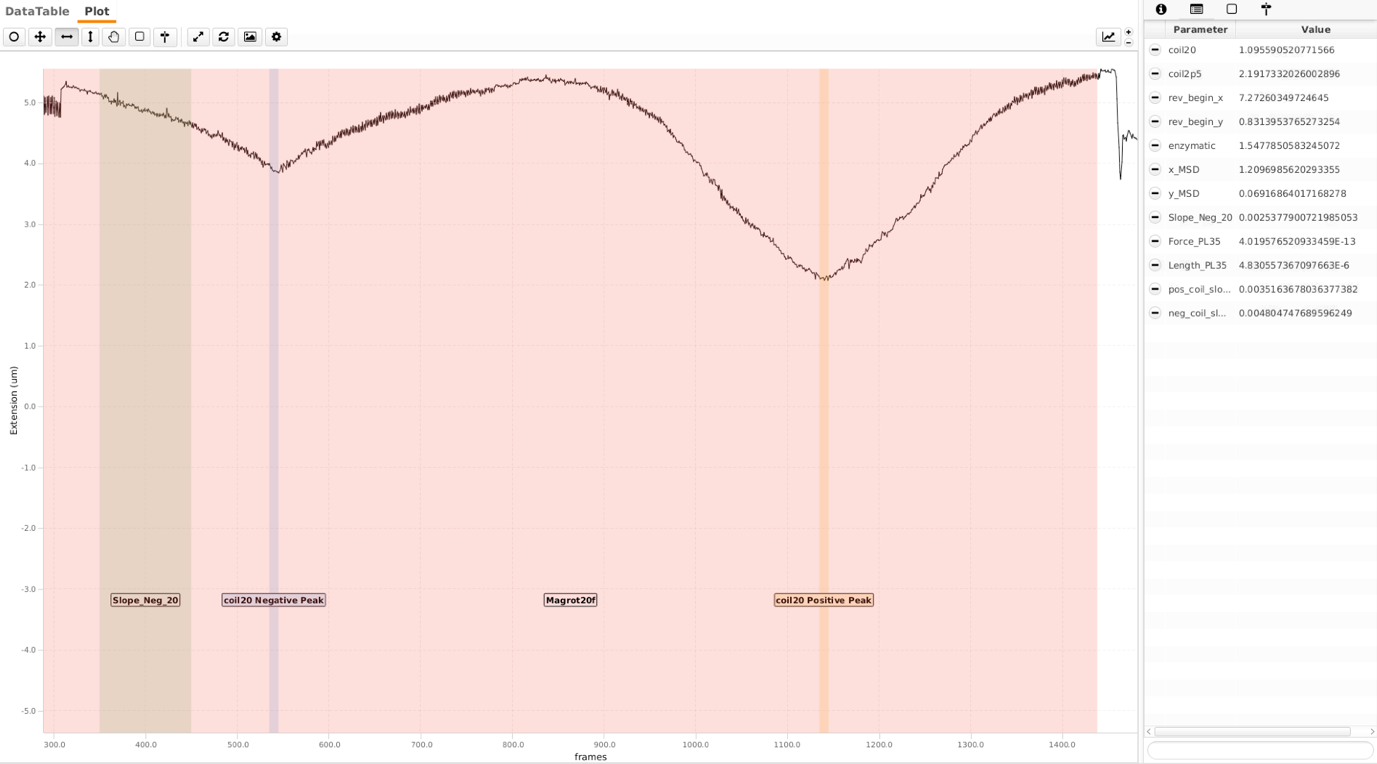
Following the reversal, magnets are rotated at 2rot/sec and high flow rate (20 μl/min - ca. 2pN) for 1200 frames to distinguish singly-tethered molecules. Two different parameters are calculated here. Here the magnets were rotated 150 turns in both positive and negative directions. ‘Magrot20f’ is the region where the magnets are rotated. ‘coil20 Negative Peak’ and ‘coil20’ positive peak are respective regions where sigma for DNA is highest for respective coilings. A single peak is observed here as DNA separates locally when negatively coiled, dissipating the torsion (see first screenshot on the bottom). This is not the case for bead that is tethered via multiple DNA molecules (see second screenshot, there are to peaks). To separate these molecules further, an additional region ‘Slope_Neg_20’ is calculated. The ‘Slope_Neg_20’ parameter is the slope of the region shown. Following thresholds are empirically chosen. The tags here are ‘doubleCoiling’ and ‘coilable20’ respectively.
//Get regions
coil20_Positive_Peak = metadata.getRegion("coil20 Positive Peak")
coil20_Negative_Peak = metadata.getRegion("coil20 Negative Peak")
//RegionDifferenceCalculatorCommand.calcRegionDifference(molecule, xColumn, yColumn, regionOne, regionTwo, parameterName)
//coil20
RegionDifferenceCalculatorCommand.calcRegionDifference(molecule, "slice", "x", coil20_Positive_Peak, coil20_Negative_Peak, "coil20")
//coilable20
if (molecule.getParameter("coil20") > coilable20LowerBound &&\
molecule.getParameter("coil20") < coilable20UpperBound) {
molecule.addTag("coilable20")
}
//Get regions
Slope_Neg_20 = metadata.getRegion("Slope_Neg_20")
//Calculate slopes
output = table.linearRegression("slice","x", Slope_Neg_20.getStart(), Slope_Neg_20.getEnd())
molecule.setParameter("Slope_Neg_20", output[2])
//doubleCoiling
if (molecule.getParameter("Slope_Neg_20") > doubleCoilingLowerBound &&\
molecule.getParameter("Slope_Neg_20") < doubleCoilingUpperBound) {
molecule.addTag("doubleCoiling")
}
Force calculation
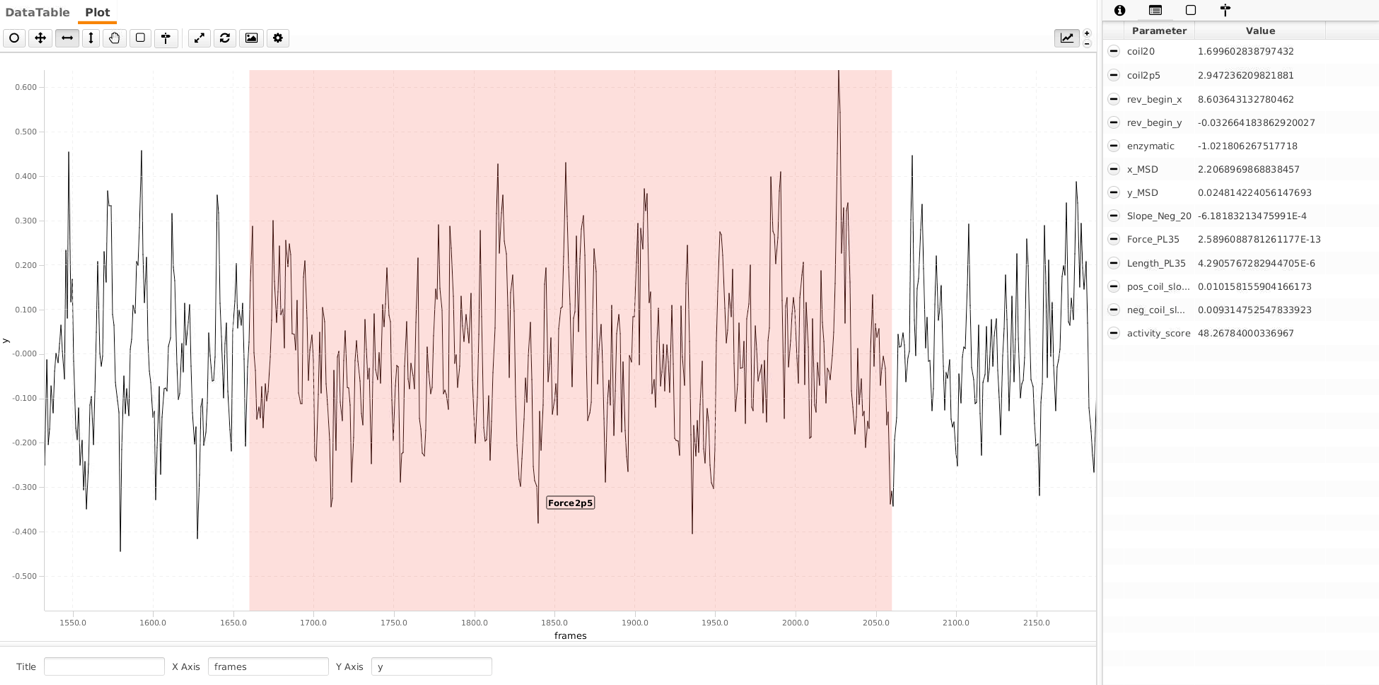
Bead fluctuations lateral to flow direction are used to numerically ascertain force and length of the DNA molecule. As only the projected length is known, DNA is assumed to have worm-like chain behavior.

The δy2 is the the mean squared displacement of the bead excursions in the y direction. They are taken from the Force2p5 region.
Other parameters such as ‘temperature’, ‘persistanceLength’, ‘contourLength’ are given in the workflow as constants.
The other two values for ‘slidingForceWindow’ and ‘stdSlidingForce’ are used to check if the bead, in the chosen, get transiently stuck. If the bead gets stuck, it registers as higher force. The sliding windows iteratively calculates mean and standard deviation of each window. If a particular window deviates more than threshold MSD, the bead is tagged as ‘stuckForce’. This also makes sense as MSD of these fluctuations are supposed to be constant throughout the region. The screenshot in the bottom shows how the region which is used to calculate the force could look like. You can see the fluctuations.
//Get regions
Force2p5 = metadata.getRegion("Force2p5")
//Force Calculation and tagging
logService.info("Calculating force...")
archive.getMoleculeUIDs().parallelStream().forEach{ UID ->
Molecule molecule = archive.get(UID)
MarsTable table = molecule.getTable()
double msdForce = table.msd("y_drift_corr", "slice", Force2p5.getStart(), Force2p5.getEnd()) //inserts different start and stop slice with each loop
msdForce = msdForce*conversionPixelToMicron*conversionPixelToMicron
double[] solution;
try {
solution = MarsMath.calculateForceAndLength(persistenceLength, contourLength, temperature, msdForce);
} catch (Exception e) {
return;
}
double force = solution[0]
double length = solution[1]
String force1 = "Force_PL35"; // make Force_2.5, Force_5 for different flow rates
String length1 = "Length_PL35"; // similarly for length
molecule.setParameter(force1, force);
molecule.setParameter(length1, length);
DoubleColumn msdColSlide = new DoubleColumn("MSDs")
double MaxMSD = 0
double MinMSD = 0
for (int row = 0; row < table.getRowCount() - slidingForceWindow; row++) {
if (table.get("slice",row) >= Force2p5.getStart() && table.get("slice",row) <= Force2p5.getEnd() && table.getRowCount() > row + slidingForceWindow) {
double msdSlideForce = table.msd("y_drift_corr","slice",table.get("slice",row),table.get("slice",row + slidingForceWindow))
if (msdSlideForce > MaxMSD)
MaxMSD = msdSlideForce
if (msdSlideForce < MinMSD)
MinMSD = msdSlideForce
msdColSlide.add(msdSlideForce)
}
}
MarsTable tempTable = new MarsTable("MSDs table")
tempTable.add(msdColSlide)
double msdSTD = tempTable.std("MSDs")
double meanMSD = tempTable.mean("MSDs")
if (msdSTD*stdSlidingForce > Math.abs(MaxMSD - meanMSD)) {
molecule.addTag("stuckForce")
} else if (msdSTD*stdSlidingForce > Math.abs(meanMSD - MinMSD)) {
molecule.addTag("stuckForce")
}
archive.put(molecule)
}
Rotation at reaction flow rate
Before the gyrase reaction, the molecule is supercoiled at reaction flow rate (which means that the force on the bead is the same during rotating the magnet in this step and also later during the gyrase reaction). This part of the trace shows the molecule being coiled negatively at first and then positively. Ultimately, the tether is left at +60 turns for the gyrase reaction. Similar to the ‘coilable20’ tag, ‘coilable2p5’ is a region difference calculation. It helps further refine finding optimal traces. The regions used for this tag are ‘coil2p5 Peak’ and ‘coil2p5 Background’.
‘Positive Coiling Slope’ and ‘Negative Coiling Slope’ regions are used as coordinate conversion tool from pixels to cycles/sec later during the gyrase reaction. Here the magnet rotation is 1 rot/sec and frame acquisition rate is 4 frames per second. Hence ‘turns_per_slice’ is 1 rot/4 frames = 0.25. ‘turns_per_cycle’ is one cycle of gyrase as it is a type II topoisomerase.
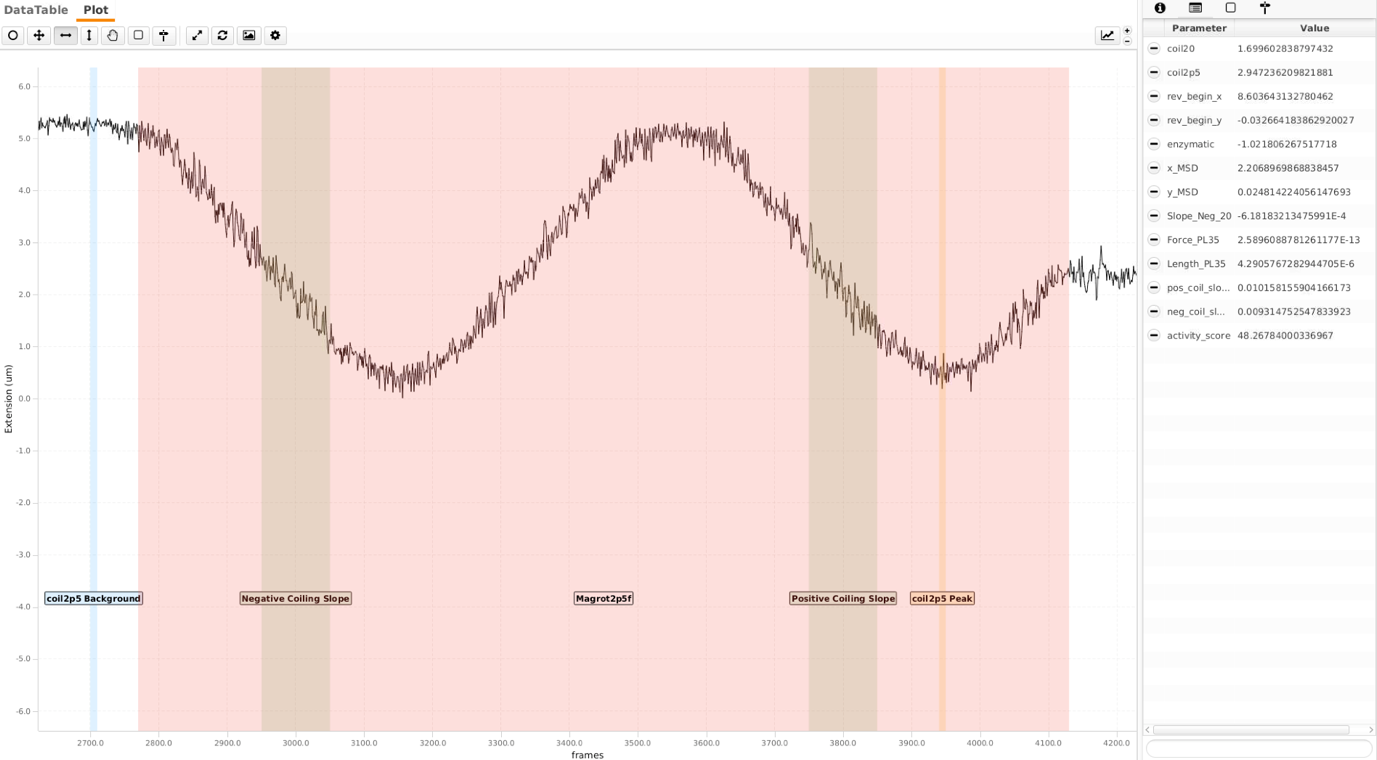

//Get regions
Negative_Coiling_Slope = metadata.getRegion("Negative Coiling Slope")
Positive_Coiling_Slope = metadata.getRegion("Positive Coiling Slope")
//Calculate poscycles and negcycles on a molecules-by-molecule basis
logService.info("Calculating molecule-by-molecule cycle numbers...")
archive.getMoleculeUIDs().parallelStream().forEach({ UID ->
Molecule molecule = archive.get(UID)
MarsTable table = molecule.getTable()
double[] pos_coils_per_slice = table.linearRegression("slice", "x_drift_corr", Positive_Coiling_Slope.getStart(), Positive_Coiling_Slope.getEnd())
molecule.setParameter("pos_coil_slope", pos_coils_per_slice[2])
if (!table.hasColumn("poscycles"))
table.appendColumn("poscycles")
for (int row = 0; row < table.getRowCount(); row++) {
double conversion = (-1)*(turns_per_slice/pos_coils_per_slice[2])/turns_per_cycle
table.setValue("poscycles", row, conversion*table.getValue("x_drift_corr", row))
}
double[] neg_coils_per_slice = table.linearRegression("slice", "x_drift_corr", Negative_Coiling_Slope.getStart(), Negative_Coiling_Slope.getEnd())
molecule.setParameter("neg_coil_slope", neg_coils_per_slice[2])
if (!table.hasColumn("negcycles"))
table.appendColumn("negcycles")
for (int row = 0; row < table.getRowCount(); row++) {
double conversion = (1)*(turns_per_slice/neg_coils_per_slice[2])/turns_per_cycle
table.setValue("negcycles", row, conversion*table.getValue("x_drift_corr", row))
}
archive.put(molecule)
})
Gyrase reaction
Final part of the trace after all the checkpoints is the gyrase reaction (shown in the screenshot below).
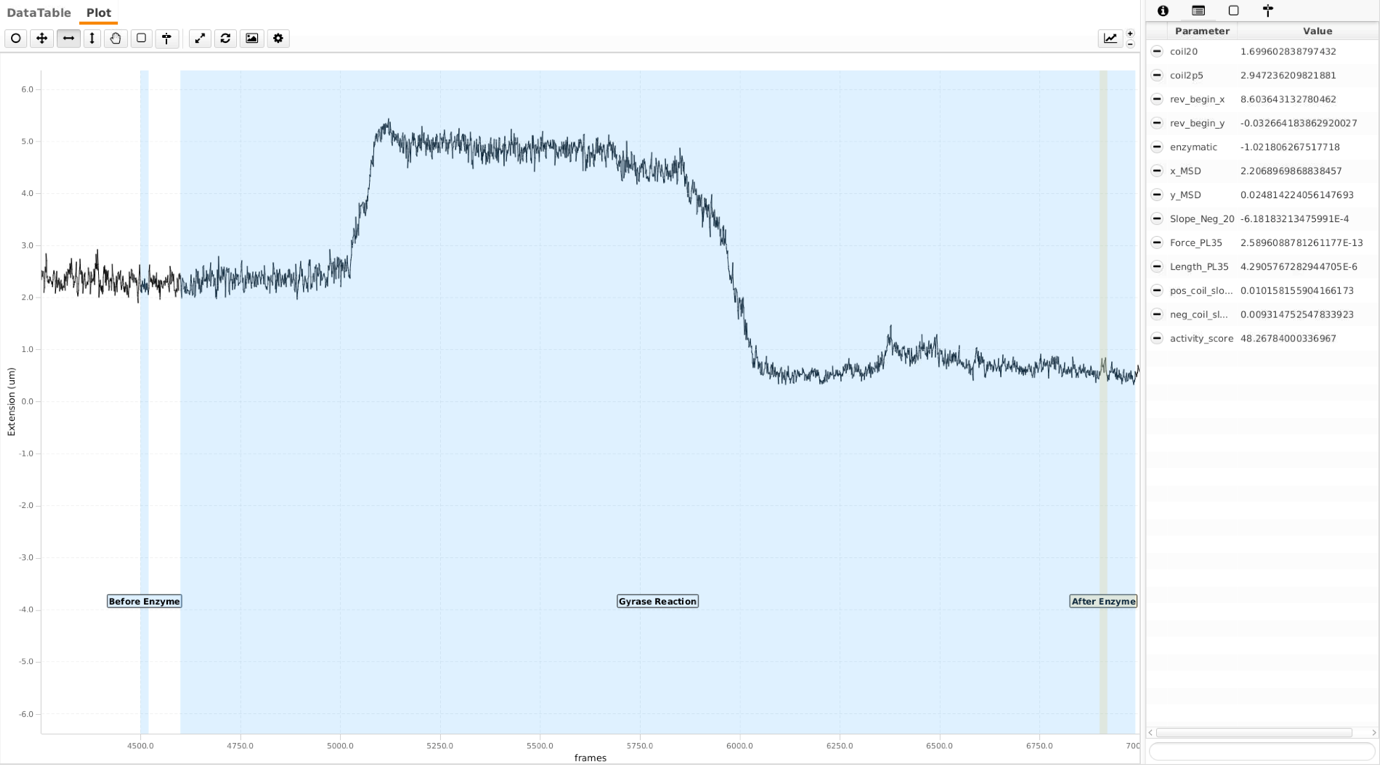
In the gyrase reaction region, there are ‘Before Enzyme’ and ‘After Enzyme’ regions. As gyrase reactions are force dependent, for any force above ca. 0.5 pN, only positive coils are resolved (chi reaction). For lower forces, both positive coils are resolved and negative coils are introduced (alpha reactions). Following thresholds tag molecules either alpha or chimol. Also, the parameter activity_score is calculated in the gyrase Reaction region.
//Calculating activity score
.forEach({ UID ->
Molecule molecule = archive.get(UID)
MarsTable table = molecule.getTable()
if (table == null)
return
double startTime = Gyrase_Reaction.getStart()
double endTime = Gyrase_Reaction.getEnd()
if (table.getValue("slice", table.getRowCount()-1) < Gyrase_Reaction.getEnd()) {
endTime = table.getValue("slice", table.getRowCount()-1) - 10
}
double gMax = -1000000
double gMin =1000000
for (int row=0; row<table.getRowCount();row++) {
double start = table.getValue("slice", row)
double end = table.getValue("slice", row)
if (start >= startTime && end <= endTime) {
double cMax = table.getValue("poscyclesG", row)
if (gMax < cMax)
gMax = cMax
double cMin = table.getValue("poscyclesG", row)
if (gMin > cMin)
gMin = cMin
}
}
molecule.setParameter("activity_score", gMax - gMin)
Additional Thresholds shown here:
//From the reversal parameters calculated
//stuckRev
if (Math.abs(molecule.getParameter("rev_begin_x")) < stuckRevThreshold &&\
Math.abs(molecule.getParameter("rev_begin_y")) < stuckRevThreshold) {
molecule.addTag("stuckRev")
}
//stuckMSD
if (molecule.hasTag("stuckRev") &&\
molecule.getParameter("x_MSD") < stuckMSDThresholdx &&\
molecule.getParameter("y_MSD") < stuckMSDThresholdy) {
molecule.addTag("stuckMSD")
}
//shortTrajectory
int deadSlice = table.getValue("slice",table.getRowCount()-1)
if (deadSlice < minLength)
molecule.addTag("shortTrajectory")
//Drift Calculator
DriftCalculatorCommand.calcDrift(archive, "stuckMSD", "x", "y", "x_drift", "y_drift", false, "mean", "end")
//Drift Corrector
DriftCorrectorCommand.correctDrift(archive, driftZeroRegionStart, driftZeroRegionEnd, "x_drift", "y_drift", "x", "y", "x_drift_corr", "y_drift_corr", false)
‘dirftZeroRegion’ is the number of frames during drift correction where coordinates are set to 0 for this reference region. ‘conversionPixeltoMicron’ is the pixels size in microns.
‘stuckRevThreshold’ uses the reversal parameters ‘rev_begin_x’ and ‘rev_begin_y’ to tag immobile beads. If there is no motion during flow reversal in either x or y coordinate these are tagged ‘stuckRev’.
‘stuckMSDThresholdx’ and ‘stuckMSDThresholdy’ parameters are calculated MSD for the whole trace for x and y respectively. Molecules within the threshold are tagged stuckMSD. These are used to calculate and correct for drift.
‘minLength’ tags molecules ‘shortTrajectory’. This filters out all the trajectories which have less frames than the provided threshold.