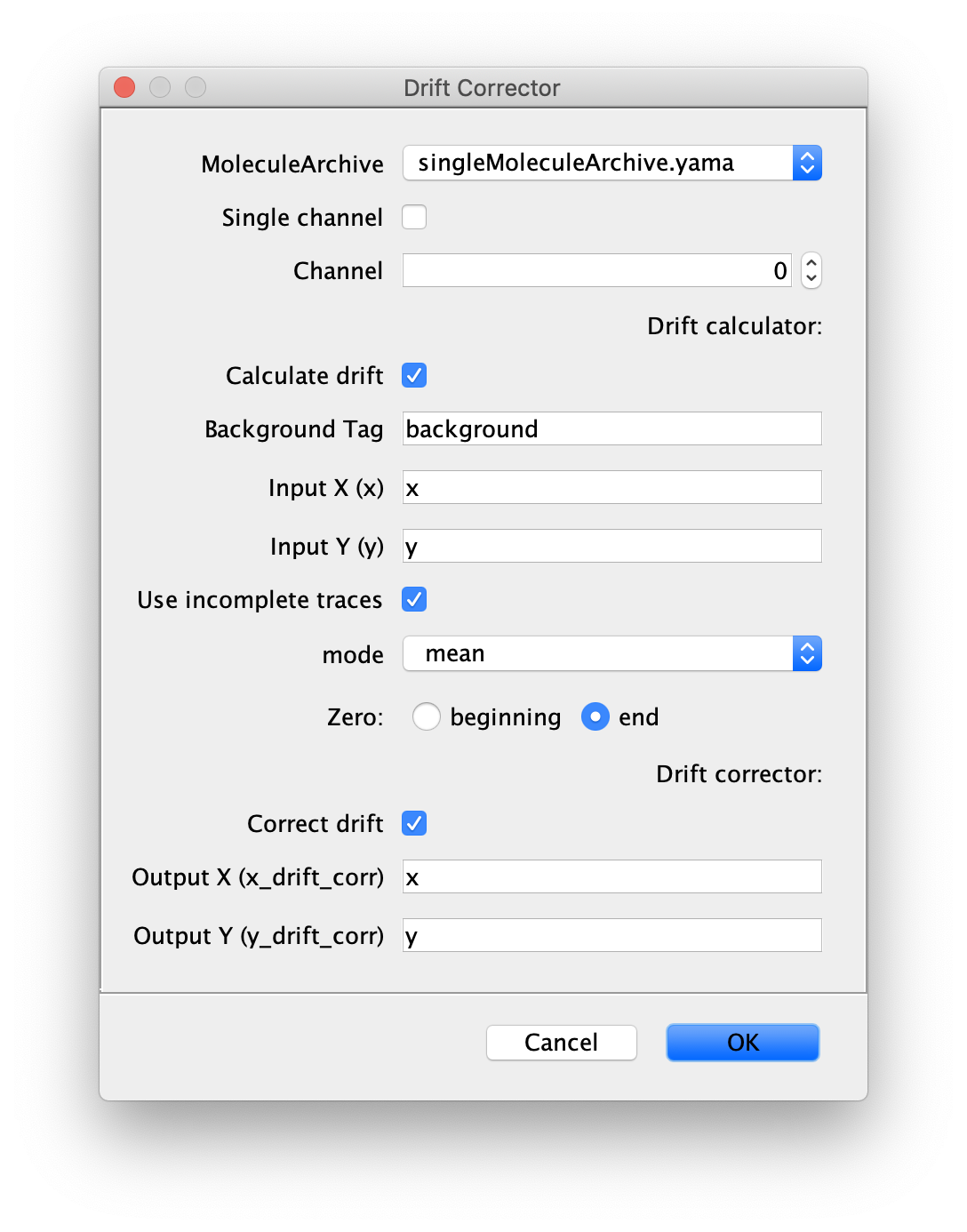
Drift Corrector
This command calculates the sample drift given a MoleculeArchive and a tag corresponding to all immobile molecules. It calculates the mean or median x and y position for each frame (T) of these immobile molecules and reports those results in the metadata specified per plane (xDrift, yDrift). If selected, the coordinates for the molecules in the MoleculeArchive will be corrected for drift by generating new columns for each molecule table listing ‘x_drift_corr’ and y_drift_corr.
Inputs
- MoleculeArchive - MoleculeArchive for which drift will be corrected. The drift will be subtracted from the x and y columns for each molecule using the corresponding drift information.
- Single Channel - Tick this box if the drift should only be calculated for all data corresponding to a certain channel. The drift correction can be executed sequentially for all channels in the archive if drift differences are to be expected between different channels.
- Channel - Select the channel for which the drift should be calculated in case the ‘single channel’ option is ticked.
- Calculate drift - Tick this box if the drift should be calculated.
- Background Tag - The tag added to all molecules that should be used to calculate background.
- Input X (x) - X-coordinates to be corrected. Usually this is just x.
- Input Y (y) - Y-coordinates to be corrected. Usually this is just y.
- Use incomplete traces - Uses all traces for the drift calculation including those that do not have all slices, which sometimes happens when peak fitting fails for a certain frame. If there are no observations for a certain slice, then NaN is used.
- mode - Select between using the ‘mean’ or ‘median’ x and y position.
- Zero - Select between treating the starting point or then end point as zero.
- Correct drift - Select if the coordinates should be corrected for drift.
- Output X (x_drift_corr) - The new output x column added to the table with the background corrected trace. Usually this is x_drift_corr.
- Output Y (y_drift_corr) - The new output y column added to the table with the background corrected trace. Usually this is y_drift_corr.
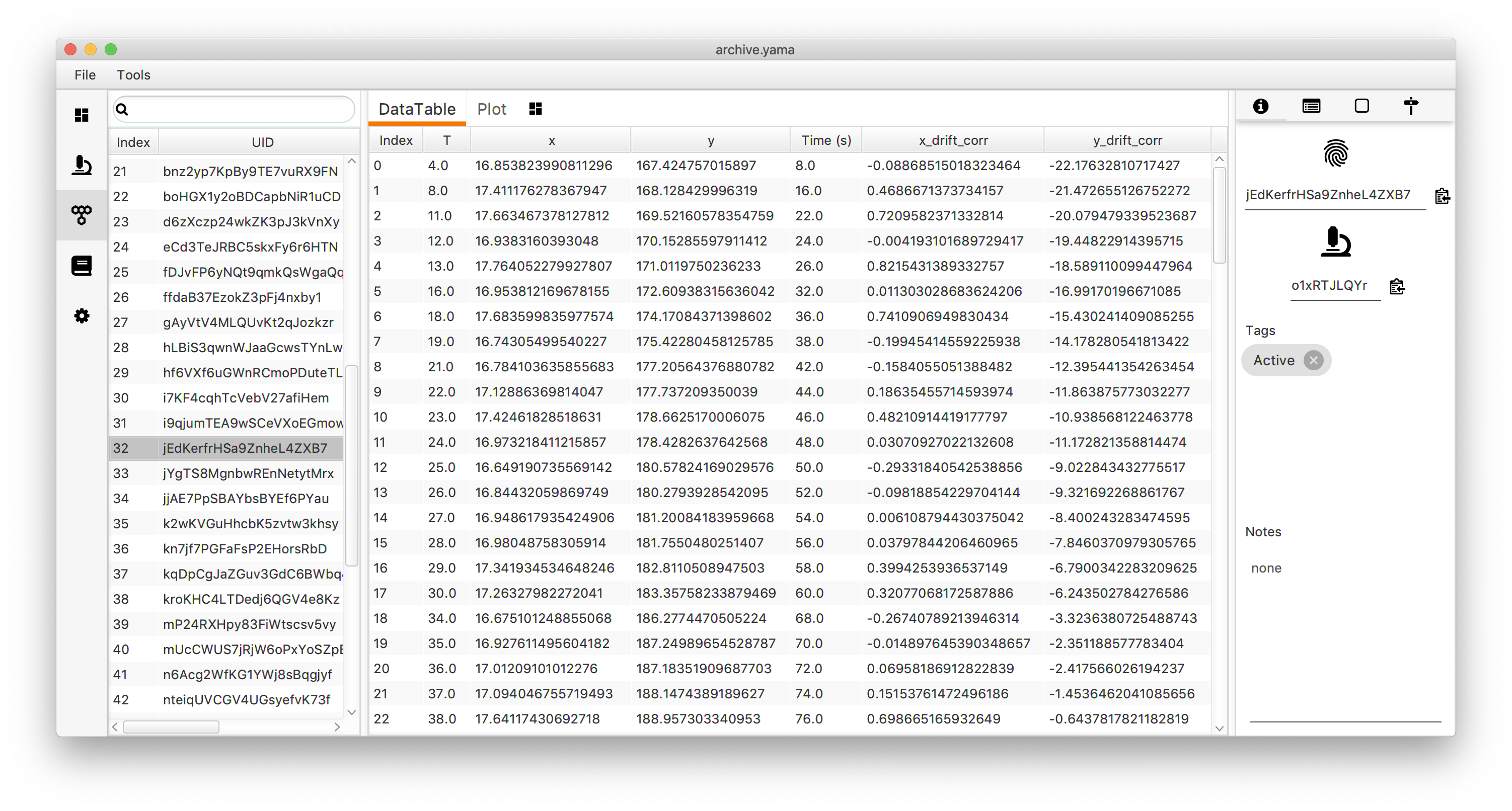
Output
- The MoleculeArchive provided as Input is modified. The drift is subtracted from the x and y columns to generate x_drift_corr and y_drift_corr. Column names will vary depending on settings…
How to run this Command from a groovy script
#@ MoleculeArchive archive
#@ ImageJ ij
import de.mpg.biochem.mars.molecule.*
//Make an instance of the Command you want to run...
final DriftCorrectorCommand driftCorr = new DriftCorrectorCommand();
//Populates @Parameters Services etc.. using the current context
//which we get from the ImageJ input...
driftCorr.setContext(ij.getContext())
//Set all the input parameters
driftCorr.setArchive(archive)
driftCorr.setSingleChannel(False)
driftCorr.setChannel(0)
driftCorr.setCalculateDrift(True)
driftCorr.setBackgroundTag("background")
driftCorr.setInputX("x")
driftCorr.setInputY("y")
driftCorr.setUseIncompleteTraces(false)
driftCorr.setMode("mean")
driftCorr.setZeroPoint("end")
driftCorr.setCorrectDrift(True)
driftCorr.setOutputX("x_drift_corr")
driftCorr.setOutputY("y_drift_corr")
//Run the Command
driftCorr.run();