Sigma Calculator
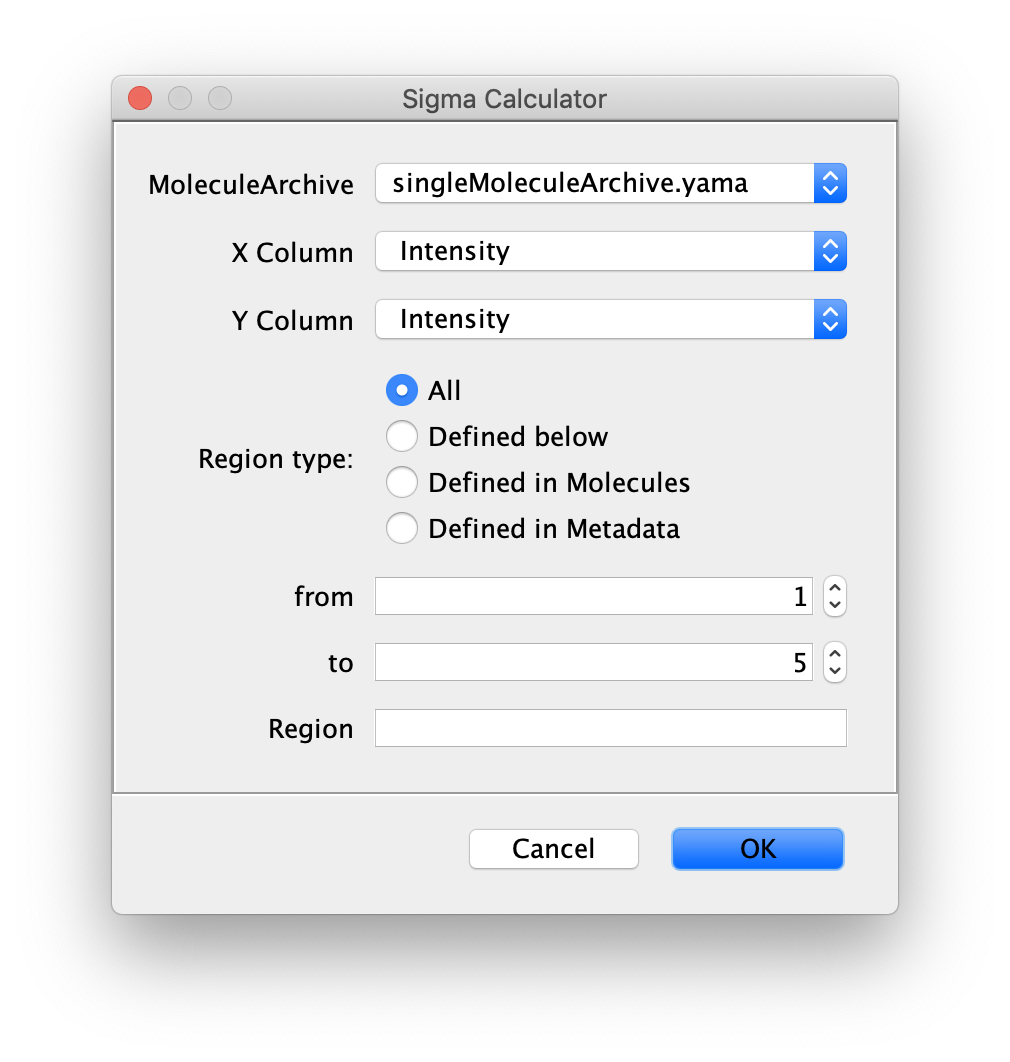
The ‘Sigma Calculator’ tool calculates the error value in a specific region of all traces in the Molecule Archive. By either specifying a region of interest manually or selecting it in the plot of a trace the sigma value for a region of interest is calculated. This error value can be used as an input in the Change Point Finder command and will automatically override a global sigma value. This gives the user the possibility to optimise the fitting process for each trace individually if desired.
Inputs
- MoleculeArchive - Select the Molecule Archive to apply the Sigma Calculator to.
- X Column - X-coordinates (f.e. time or frame).
- Y Column - Y-coordinates (axis of movement).
- Region - Select which region of the trace to use in the calculation: all slices, defined below, defined in Molecules or defined in Metadata.
When selecting the option ‘Defined below’:- from - Start frame (T) number.
- to - End frame (T) number.
- Region - Enter region name to use that is defined in the Archive.
Output
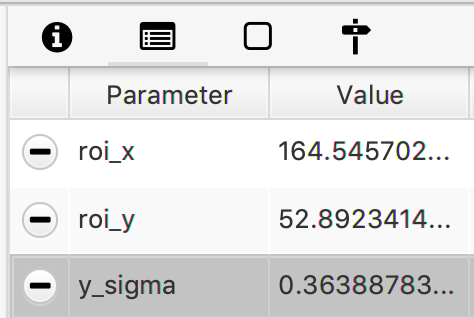
- y_sigma - The value of the calculated error at the specified settings as a parameter to each Molecule entry.
How to run this Command from a groovy script
#@ MoleculeArchive archive
#@ ImageJ ij
import de.mpg.biochem.mars.molecule.*;
//Make an instance of the Command you want to run
final SigmaCalculatorCommand sigCalc = new SigmaCalculatorCommand();
//Populates @Parameters Services etc. using the current context which we get from the ImageJ Input
sigCalc.setContext(ij.getContext());
//Set all the input parameters
sigCalc.setArchive(archive);
sigCalc.setXcolumn("T");
sigCalc.setYcolumn("y");
sigCalc.setRegionType("All");
sigCalc.setFrom("1");
sigCalc.setTo("5");
sigCalc.setRegionName("test");
//Run the Command
sigCalc.run();