Single Change Point Finder
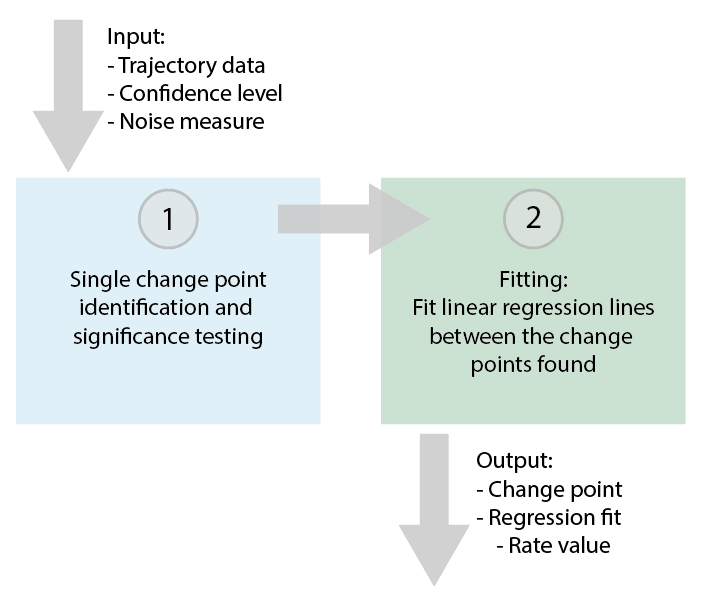
The single change point finder tool allows for the identification of one change point in the trace in the Molecule Archive. This can be of use if a bleaching step (a stepwise drop in the intensity vs. time plot indicating the bleaching of a fluorophore) has to be identified, or when the kinetics of the process studied only involve one transition.
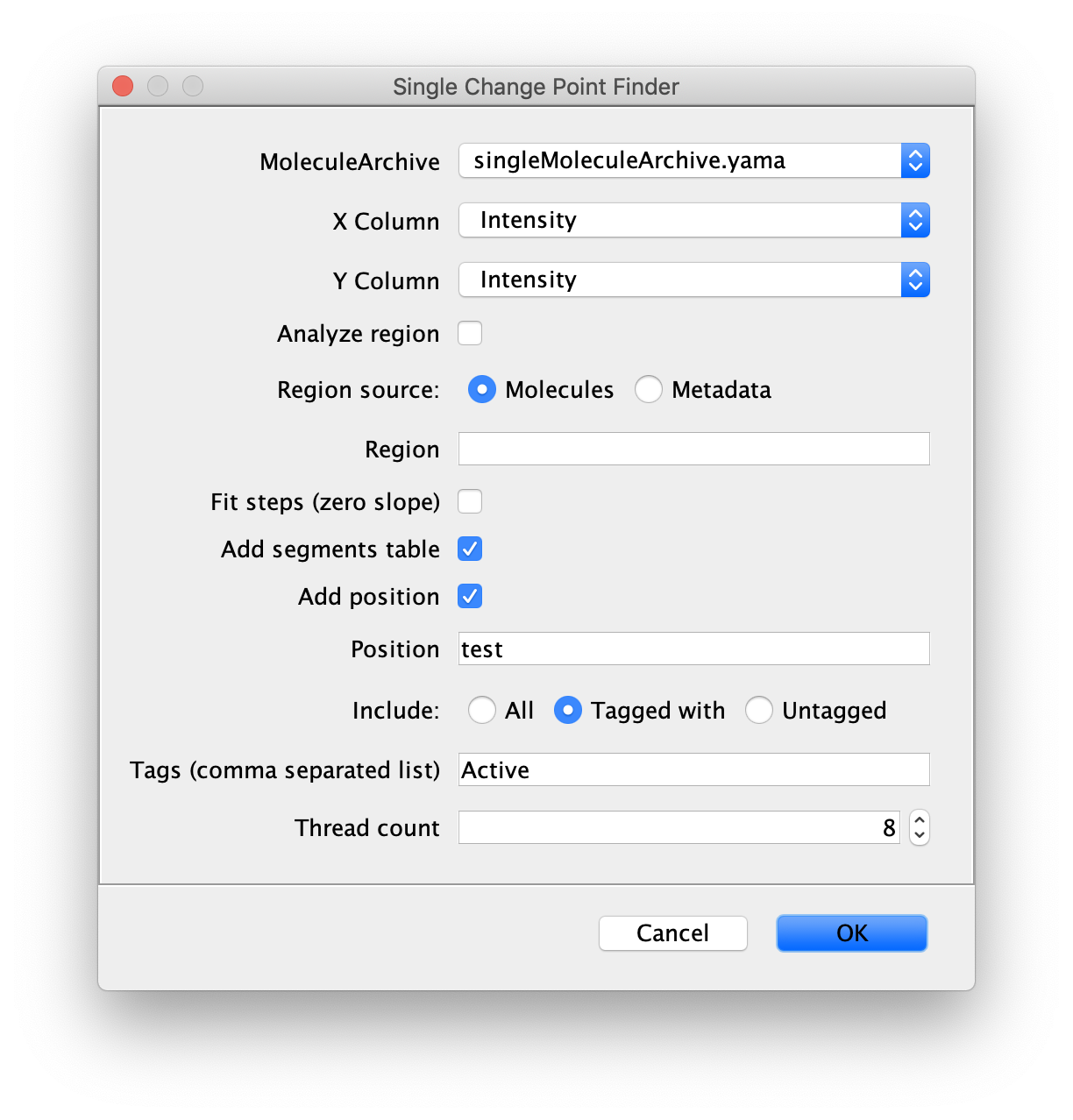
Inputs
- MoleculeArchive - Molecule Archive to find the single change point for.
- X Column - X column input (f.e. time).
- Y Column - Y column input (axis along which displacement takes place).
- Analyze region - Tick this box if only a specified region of the trace should be analyzed.
- Region source - Location of the defined region to use: Molecules or Metadata.
- Region - Name of the region to use.
- Fit steps (zero slope) - Tick box in case only lines with slope = 0 should be fitted. Use this option to for example investigate step-wise bleaching traces.
- Add segments table - Generate a segments table as output of this command. This table contains the coordinates of the change point, the slopes and corresponding errors.
- Add position - Show the position of the identified change point with an indicator in the plotted trace.
- Position - Name for the position.
- Include - Choose to include ‘all’, ‘tagged with’ or ‘untagged’ molecules in the analysis.
- Tags (comma separated list) - List of tags to base on which traces are included in the analysis.
- Thread count - Determines how much computing power of your computer will be devoted to this calculation. A higher thread count decreases computing time.
Output
- Segments table - If the ‘Add segments table’ input is enabled, a segments table is generated for each analyzed molecule containing the coordinates of the identified change point, the calculated slopes, intercepts and corresponding error values.
- Position - If the ‘Add position’ input is enabled the position of the identified change point is shown with a position marker in the plotted trace.
How to run this Command from a groovy script
#@ MoleculeArchive archive
#@ ImageJ ij
import de.mpg.biochem.mars.molecule.*;
//Make an instance of the Command you want to run
final SingleChangePointFinder scpCalc = new SingleChangePointFinder();
//Populates @Parameters Services etc. using the current context which we get from the ImageJ Input
scpCalc.setContext(ij.getContext());
//Set all the input parameters
scpCalc.setArchive(archive);
scpCalc.setXcolumn("T");
scpCalc.setYcolumn("y");
scpCalc.setAnalyzeRegion(False);
scpCalc.setRegionSource("Molecules");
scpCalc.setRegion("name");
scpCalc.setFitSteps(False);
scpCalc.setAddSegmentsTable(True);
scpCalc.setAddPosition(True);
scpCalc.setPosition("test_name");
scpCalc.setIncludeTags("Active");
//Run the Command
scpCalc.run();