Mars workflow example for static smFRET
In this example, a Mars workflow for the analysis of smFRET (single-molecule Förster Resonance Energy Transfer)1, 2, 3 is presented. This popular single-molecule technique is used extensively to investigate the behavior of bio-macromolecules like proteins, RNA, and DNA. Some examples of important processes that have been studied using smFRET are: the structural organization of bacterial RNA polymerase 4, protein-protein complex formation in neurotransmission pathways 5, dynamics of multi-domain proteins like Hsp90 in solution 6, protein folding upon ligand interaction 7, and studies of the cAMP/PKA pathways 8.
Mars offers a modular open source option for smFRET analysis with a focus on reproducibility and transparency of the analysis process. In this example, the dataset of the benchmark study published by Hellenkamp et al. 9 is analysed and compared. The example starts with localization and integration of fluorescent molecules and ends with the calculation of the FRET distributions E and S.
Table of contents
- The FRET Sample Design and Mars Analysis Process
- Data preparation
- Localization of Peaks and Intensity vs. T traces
- Data Analysis and Corrections
- Plotting & Data Exploration in Python
- Conclusion: Global Analysis Outcomes and Comparisons to Literature
- References
- Troubleshooting
The FRET Sample Design and Mars Analysis Process
Sample Design
In the experiment as designed by Hellenkamp et al. 9 the distance between two fluorophores (a donor and an acceptor) on a DNA molecule is probed. Two different samples (figure 1: 1-lo and 1-mid) are measured with a different interfluorophore distance as indicated in the figure. Since the FRET efficiency (E) is inversely correlated to the distance between the fluorophores, a different E value is to be expected for each molecule type. The fluorescence of both samples was measured using both TIRF and confocal single-molecule fluorescence set-ups after which the FRET parameters E and S were determined. In this example, the 1-lo dataset recorded on a TIRF microscope set-up will be analysed. The raw dataset can be downloaded here. More information about data aquisition and sample specifics can be found in the original publication.
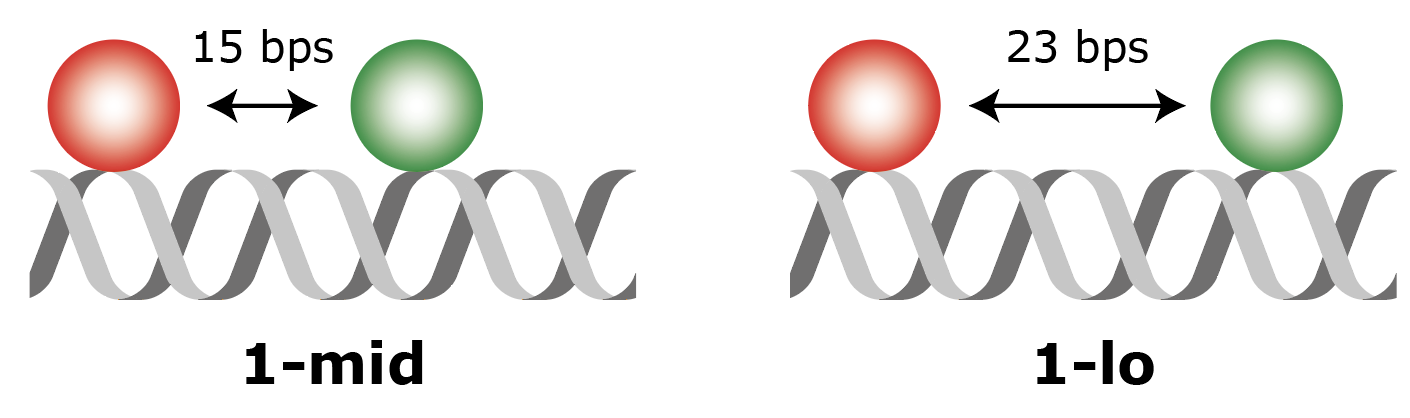
Dataset Characteristics
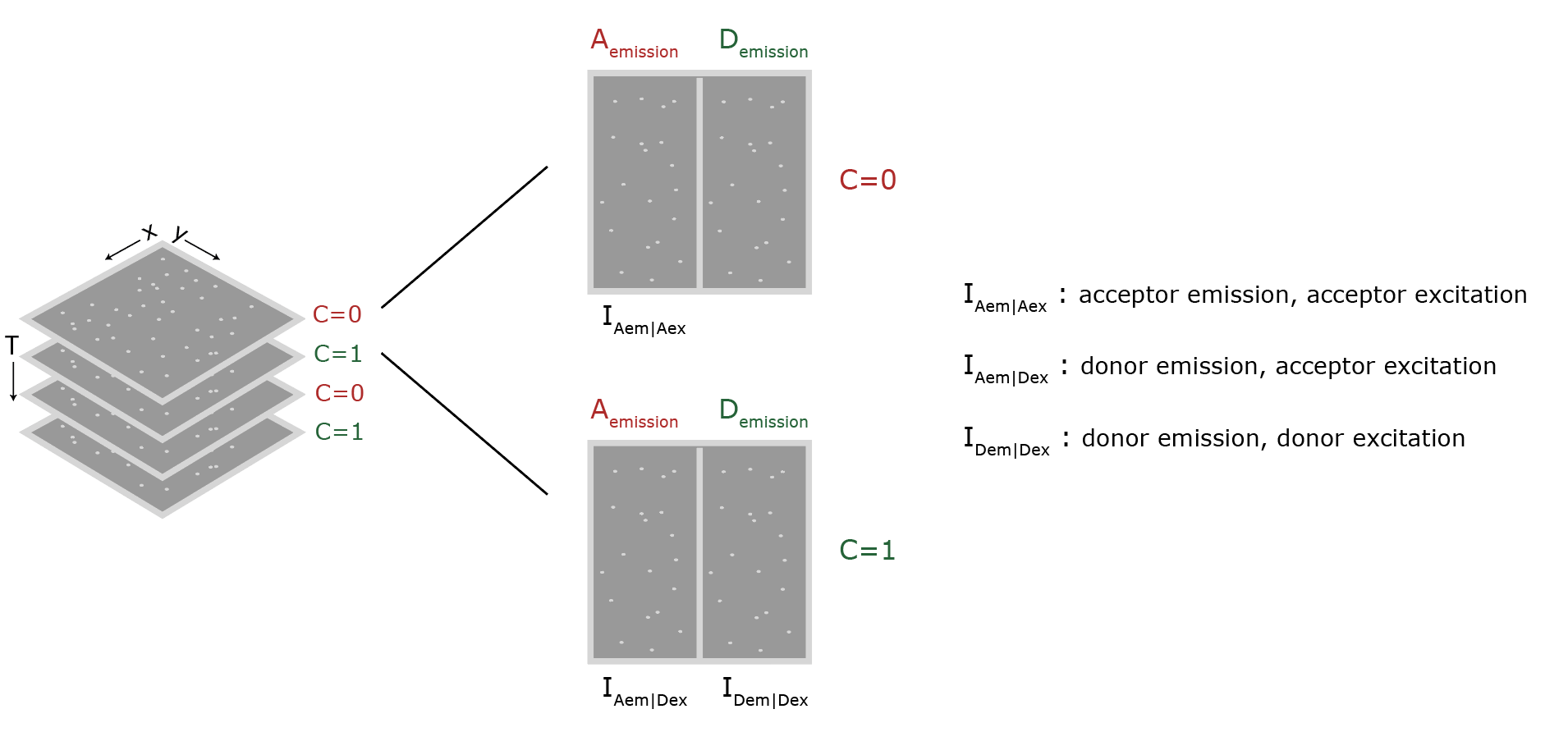
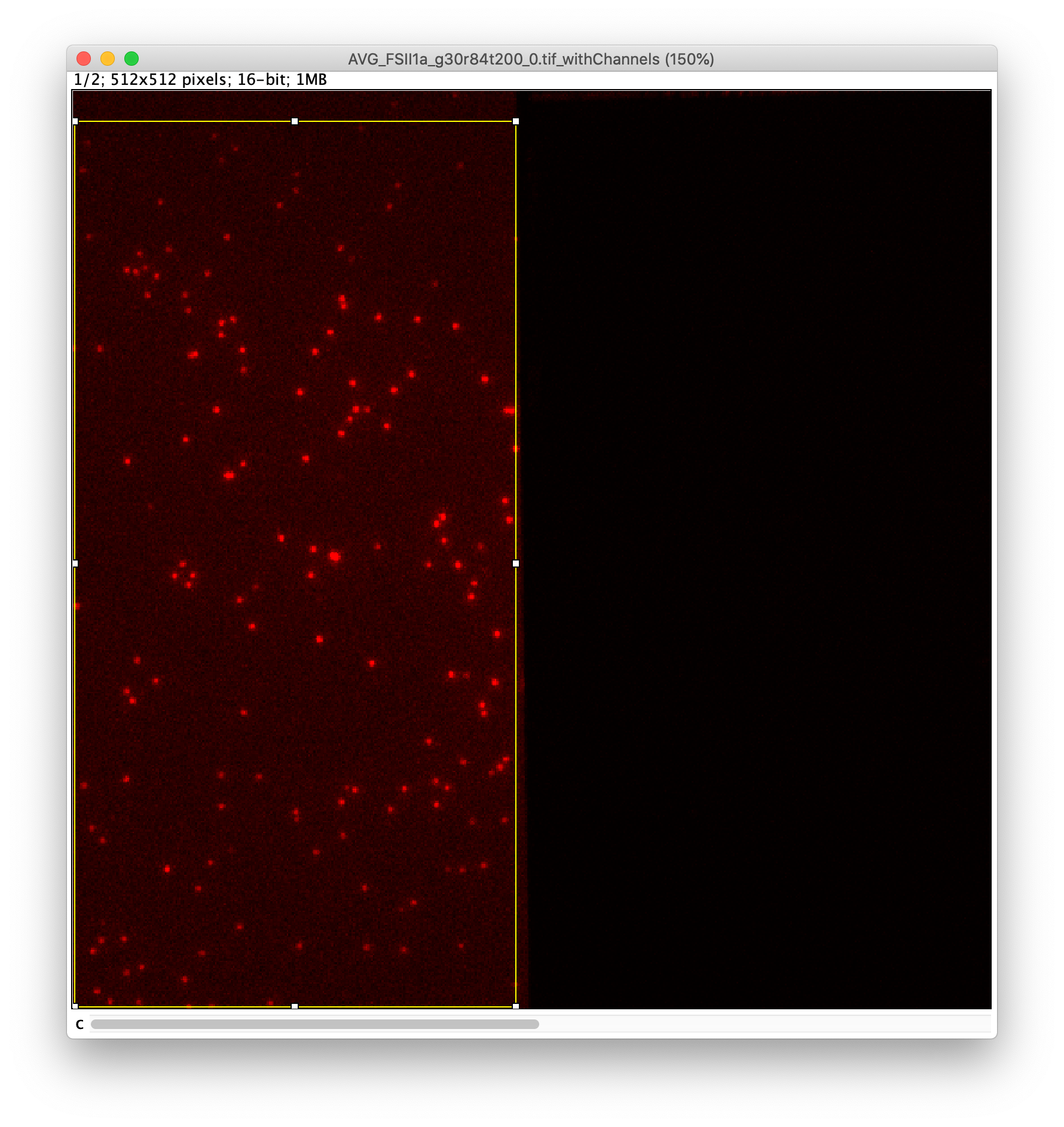
The data as provided by Hellenkamp et al. 9 was recorded with a TIRF microscope setup equipped with dual view collection. The detection area of the camera is split in half, each half displaying the signal after a different wavelength filter. In this way emission can be measured for two wavelengths at the same time and signal correlation is possible. In practice, for this dataset, this means that red emission is collected and shown on the left half, and the green emission on the right half of the window (figure 3). The excitation color alternates between red (C=0) and green (C=1) such that the different channels and split orientation give the peak intensities as denoted below each half by Iemission|excitation. These intensities are integrated during analysis and after correction lead to the final E and S values.
Comment: Some microscopes are equipped with two cameras and collect FRET images simultaneously by splitting so that the donor signal goes to one camera and acceptor signal to the other. These types of datasets also can be analyzed with this Mars workflow. We suggest combining the videos collected by the two cameras side-by-side into a single artificial dualview. This can be accomplished very easily in Fiji by opening both videos and using the combine command under Image>Stacks>Tools>Combine. This approach depends on the two cameras collecting the same number of images at the same time. Once the two videos are combined into one, this workflow can be followed exactly. This approach also ensures proper registration between the cameras that might not be perfectly aligned with sub-pixel accuracy.
The Analysis Procedure
This example was developed in a modular fashion to illustrate how workflows can be built using Mars and to simplify the process of customization for processing other FRET datasets. The workflow has two major phases. In the first phase, displayed below on the left, the emission peaks from the molecules are located and fluorescence intensity is integrated to generate Molecule Archives for donor only (DO), acceptor only (AO), and FRET populations. These are merged into one Molecule Archive that is used in Phase II, displayed on the right, where a sequence of groovy scripts provided in the mars-tutorials repository are used to process the merged Molecule Archive. This includes identification of bleaching positions, scoring intensity traces, and calculating correction factors.
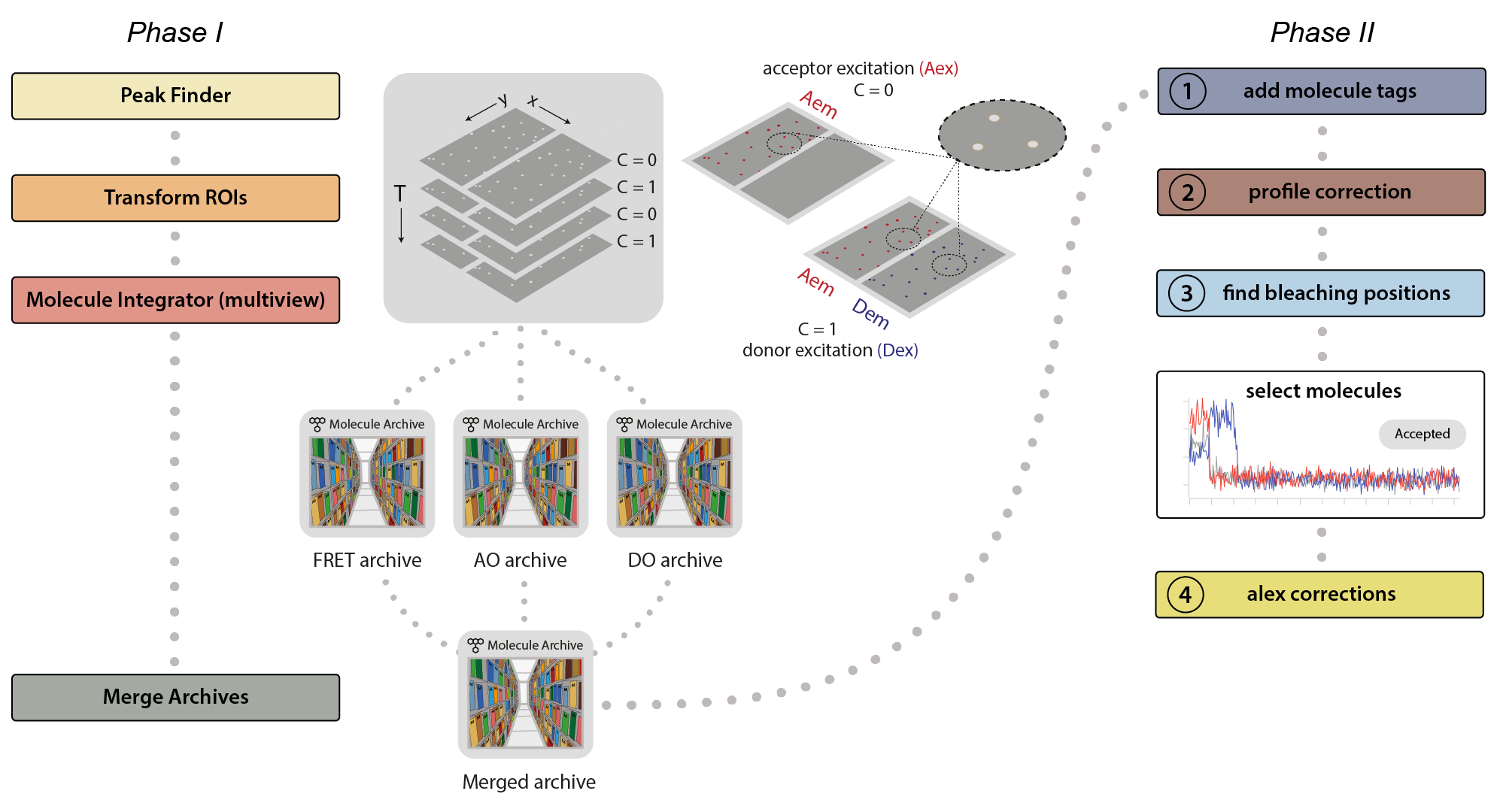
Phase I: Locate molecules and integrate fluorescence
- Identify the peaks (molecule locations) in the left or right regions.
- Correlate the peaks in the left and right regions to find which molecules have only red emission (acceptor only, AO), only green emission (donor only, DO), or emission from both dyes (FRET).
- Extract the intensity (I) vs. time (T) traces of all molecules and store this information in a Molecule Archive.
- Tag the metadata record of each Molecule Archive with DO, AO, or FRET depending on the molecules selected and integrated.
- Merge all three of the Molecule Archives (DO, AO, and FRET) in preparation for Phase II.
Phase II: Corrections, E and S calculation, kinetic analysis
- Run the add molecule tags groovy script. This script adds the tags on the metadata records (DO, AO, and FRET) to the corresponding molecule records.
- Run the profile correction groovy script. This script corrects for differences in the beam intensity across the field of view.
- Run the find bleaching positions groovy script. This script adds the donor and acceptor bleaching positions (T) to the molecule records. Evaluate the intensity traces and add the Accepted tag to passing molecules.
- Run the alex corrections groovy script. This script calculates all alex correction factors to generate the fully corrected I vs. T traces as well as the FRET efficiency (E) and stoichiometry (S) values.
The template for a custom window containing buttons for all the commands and scripts used in the FRET workflows was made using the Fiji ActionBar plugin and is provided in the mars-tutorials repository. Follow the steps in the FRET Power Tools ActionBar tutorial to setup this helpful button window in your local Fiji to simplify and speed up the workflow.
Final distributions and figures are then generated using the static FRET example jupyter notebook provided in the mars-tutorials repository. This notebook requires the file path to the final Molecule Archive generated from the scripts above as input.
Data preparation
Opening and Converting the Video
First, download the dataset, specifically the files named: ‘1_lo_TIFF.zip’, ‘1_med_TIFF.zip’ & ‘Calibration_Files_for_TIFFs.zip’.
To prepare the video for easy analysis, turn SCIFIO on in Fiji (Edit>Options>ImageJ2 tick the box) and open the first video (FSII1a_g30r84t200_0.tif) located in the ‘1_lo_TIFF.zip’ folder. The original video does not contain specific channel information but simply displays the results of the different excitation colors in an alternating fashion. To convert the video to have channel information, run the conversion script. For information on how to run a script in Fiji please read this tutorial. If the script ran correctly, a new window should have opened showing a video with the channels represented in red and green.
Calculation of the Affine2D Matrix
Next, to prepare for the data analysis, an Affine2D conversion matrix needs to be calculated. This matrix is used to correlate the peak position as found on one half of the split view to the corresponding position on the other half of the split view. More information about this calculation can be found in the Affine2D tutorial. Either follow along in the next steps to calculate the matrix or use the result from the calculation as provided in the table below.
To calculate the Affine2D matrix open the file ‘calib20140402_0.tif’ from the calibration folder ‘Calibration_Files_for_TIFFs.zip’. This file shows a recording of a bead sample that fluoresces in multiple colors upon excitation with a wide variety of wavelengths. In this way, matching of the positions in both halves of the split view is possible. To do so, select a time point of choice and duplicate the frame twice (Image>Duplicate). This will create two images of the video at the same time point. Rename the images ‘left’ and ‘right’ and in each image black out the respective halve of the view by selecting that region with the square ROI tool and pressing delete. This will result in a set of images resembling the ones in the screenshot below.
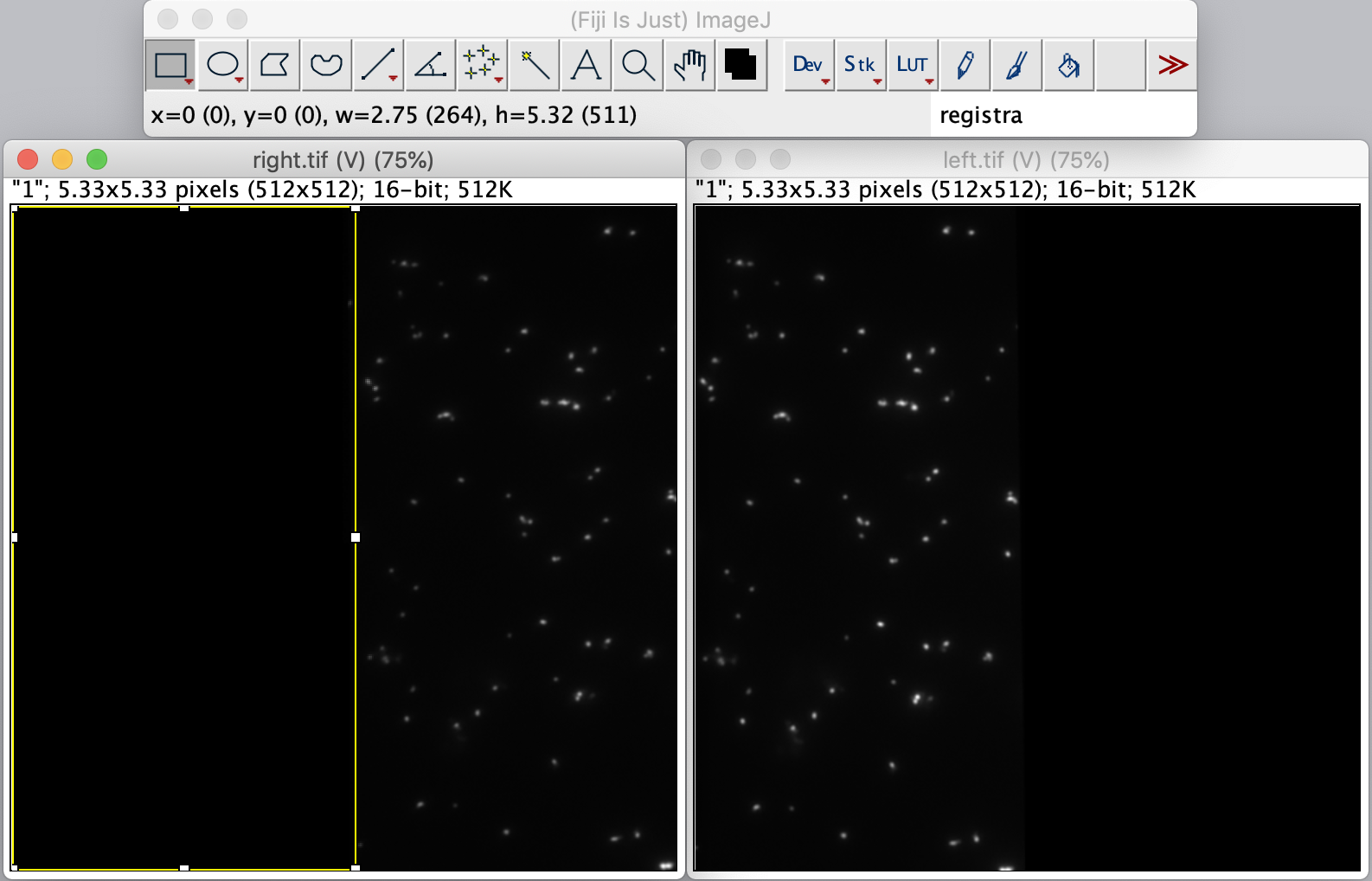
Next, open the registration tool (Plugins>Registration>Descriptor-based registration (2d/3d)) and select ‘left’ as the first image, ‘right’ as the second. Press OK and supply the settings as shown below in the next dialoge. After pressing OK, adjust the sigma and threshold values accordingly such that the peaks in the image are identified and press done. Use the default Gaussian fit parameters in the next window and press OK. This should open both an image displaying the overlap as well as a log window that displays the actual Affine2D matrix. Save the values of this matrix into a text document.
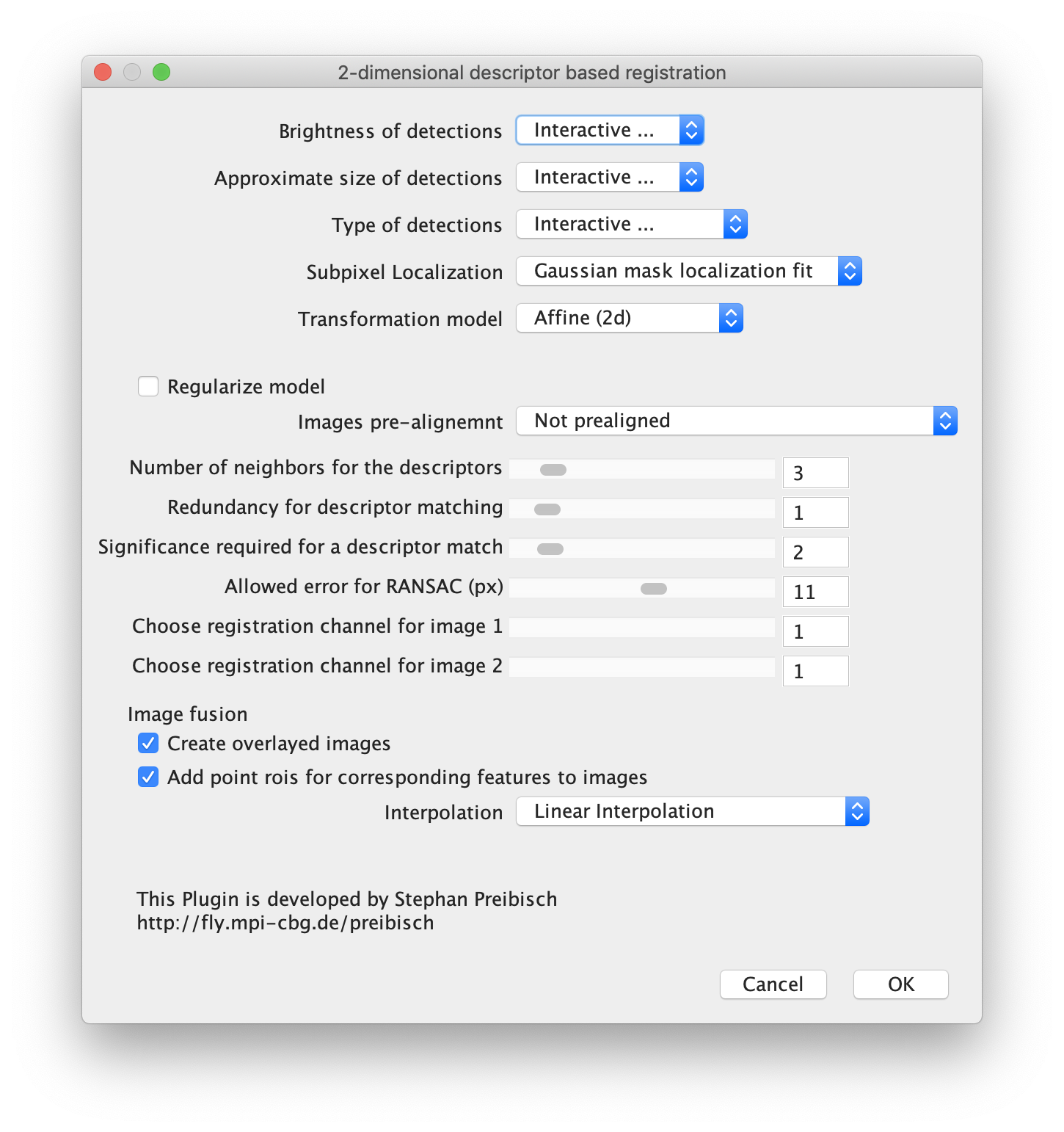
The following values were found: (note that matrices calculated at a different T or with different settings can result in slightly different values)
| Left to Right matrix | |
|---|---|
| m00 | 0.986 |
| m01 | 0 |
| m02 | 268.639 |
| m10 | 0 |
| m11 | 0.993 |
| m12 | 1.170 |
Table 1: Affine2D conversion matrix values
Find and integrate molecules
To calculate all FRET parameters and correction factors three different populations in the sample are of interest: the acceptor only (AO) population, the donor only (DO) population and the FRET population. To facilitate easy data analysis an archive is created for each of these populations separately. These are then later on merged together while ensuring the metadata records for each of these archives is tagged appropriately so molecules are correctly tagged. These tags are used when calculating the correction factors.
All three archives are created following the same workflow: peak identification, ROI transformation to the other halve of the split view, ROI filtering and finally molecule integration to obtain intensity vs. T traces in the Molecule Archive. Below, the workflow to create the FRET archive is shown with screenshots. The procedure is repeated with a few differences described for the creation of the AO and DO Molecule Archives.
The FRET Archive
Find the peaks in the red channel (C=0, left)
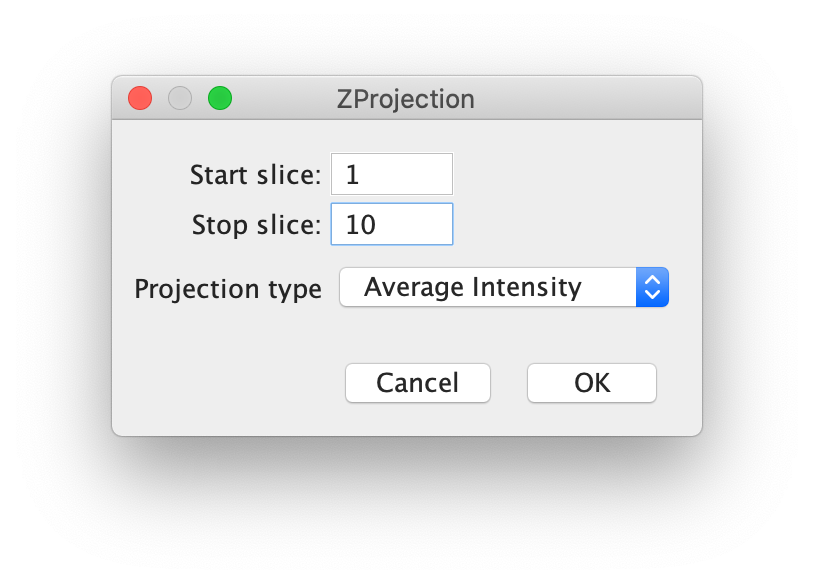
First, for a better fit performance, do a z-projection of the first 10 time points (slices in the dialog as seen below) with Projection type set to ‘Average Intensity’ yielding an average image (Fiji>image>stacks>Z project…). Use this image to find the coordinates of the peaks.
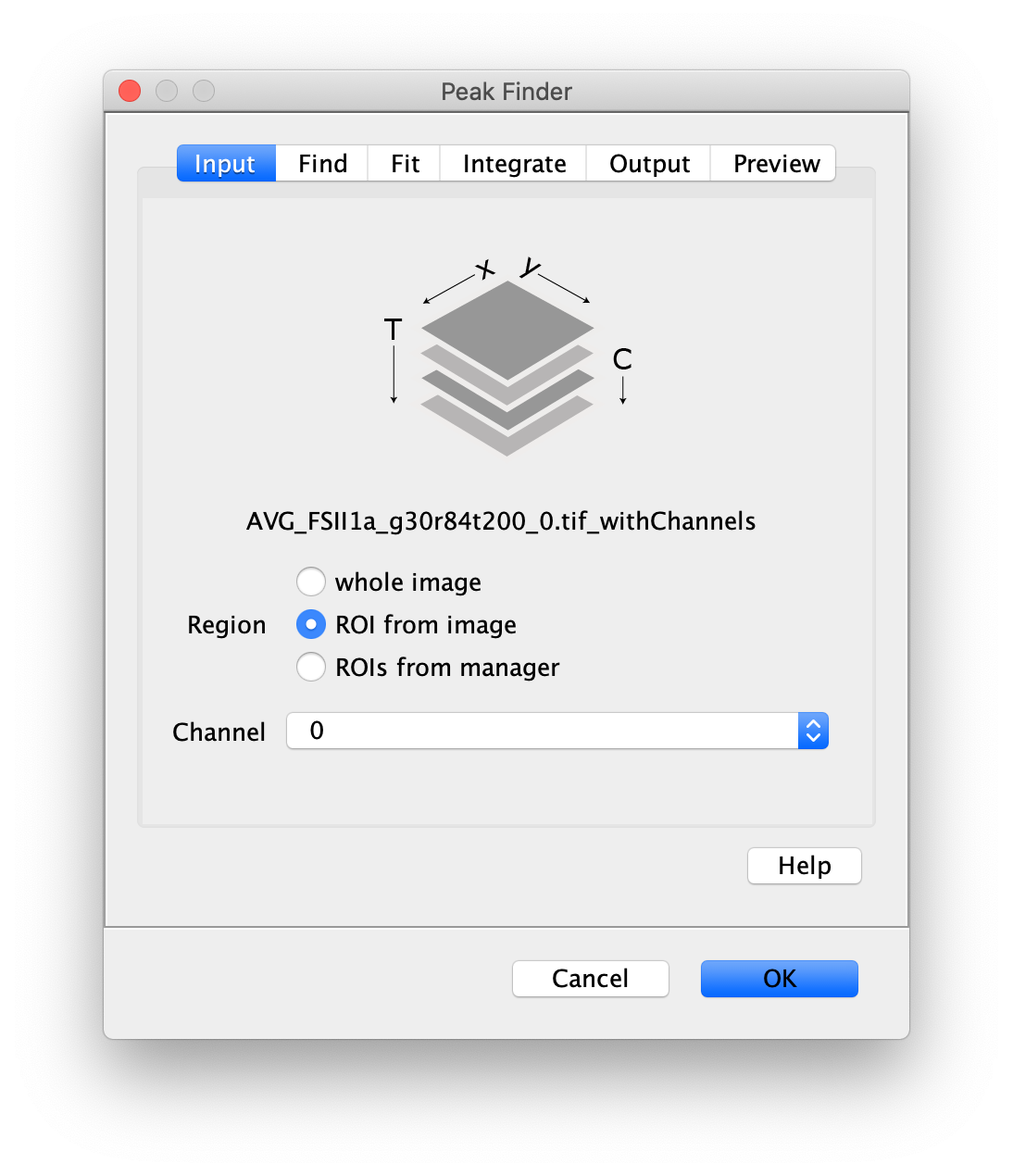
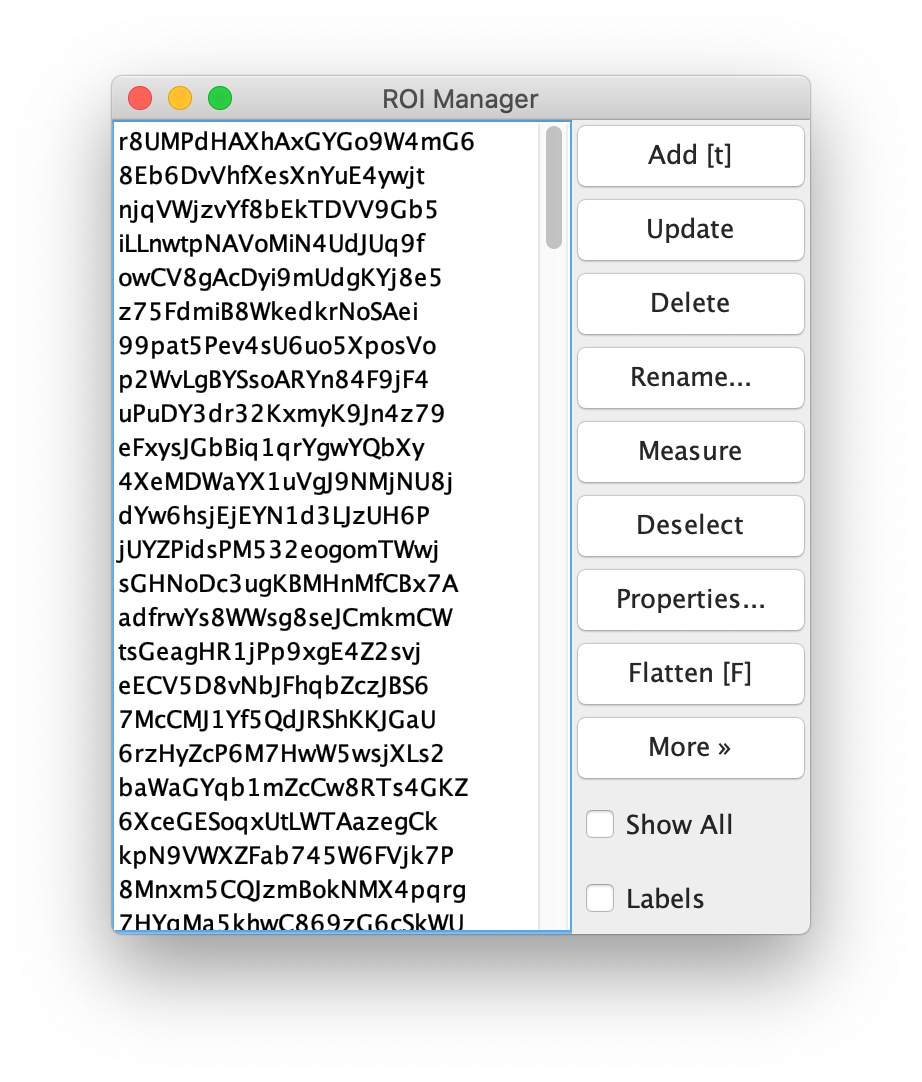
To find the peaks in the left part of the split view first select this part of the screen with the box ROI tool, do this for channel 0, and open the Peak Finder command (Plugins>Mars>Image>Peak Finder). Apply the settings as shown below and check if the peaks are identified correctly using the preview tab. If the correct settings are applied the peaks should have an identification marker (circle or point) on them. Press ok to apply the settings and add the ROIs of the identified peaks to the ROI Manager. The ROI Manager will open and will display the peaks listed by their UID in the ROI Manager.

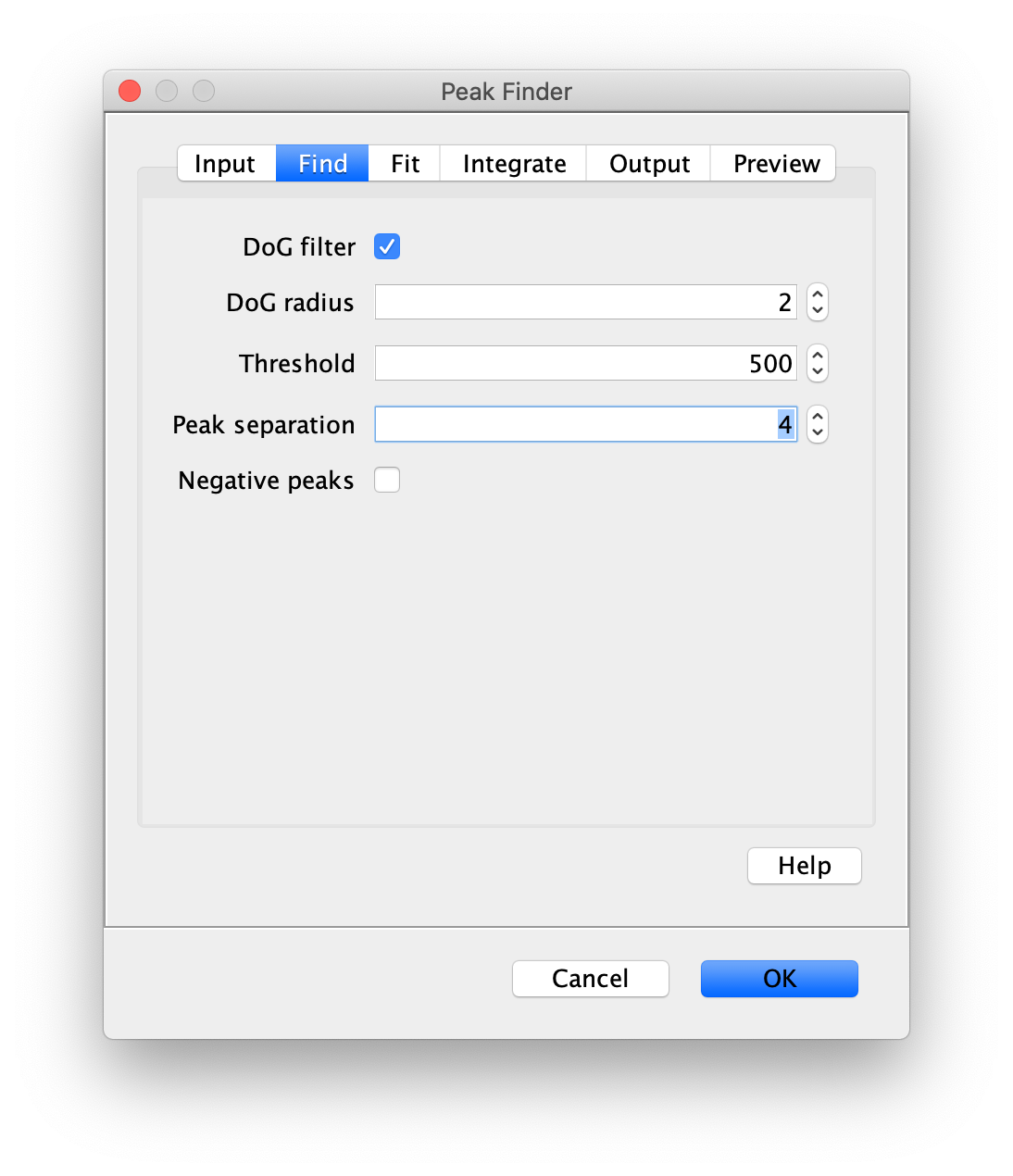
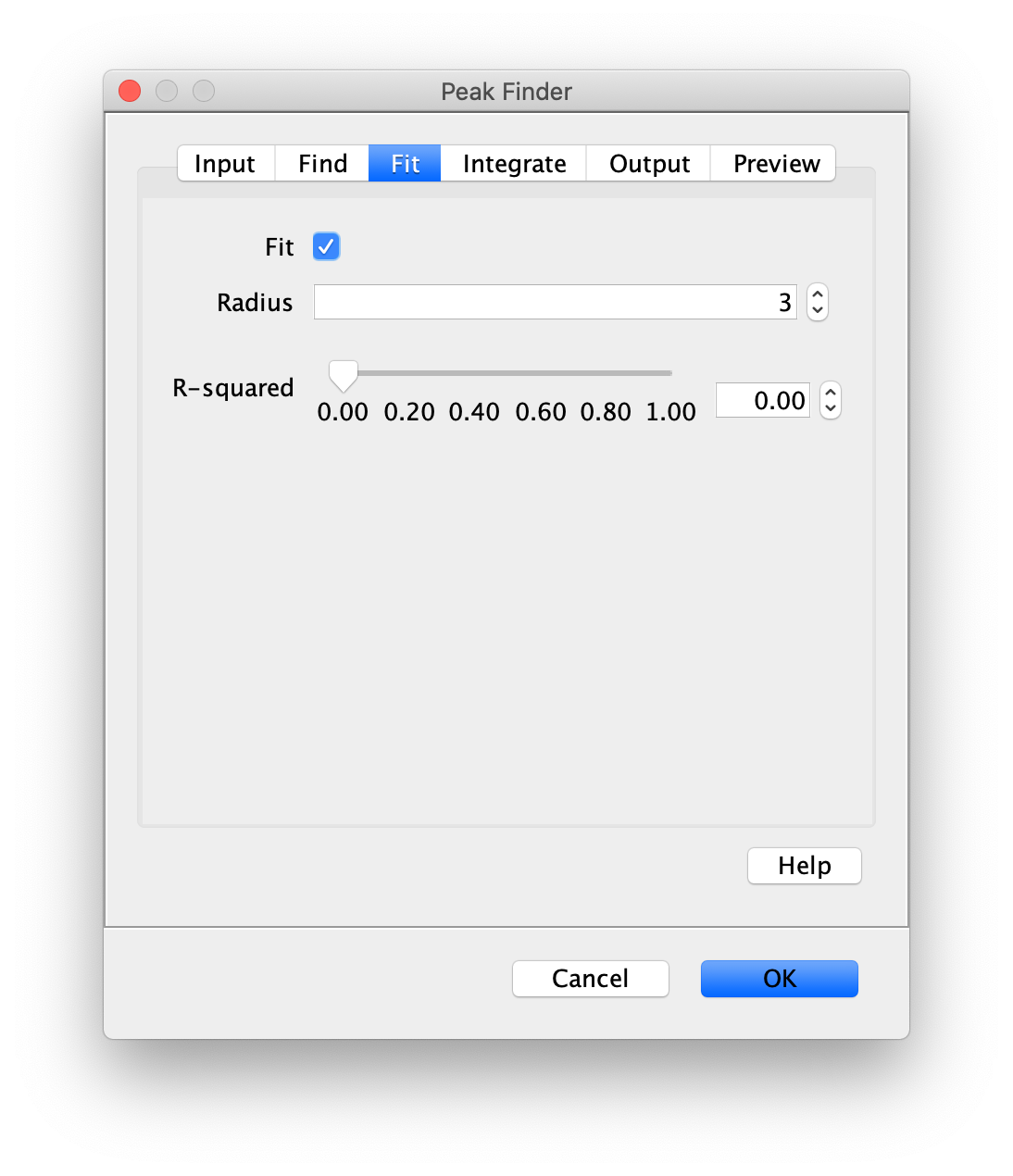
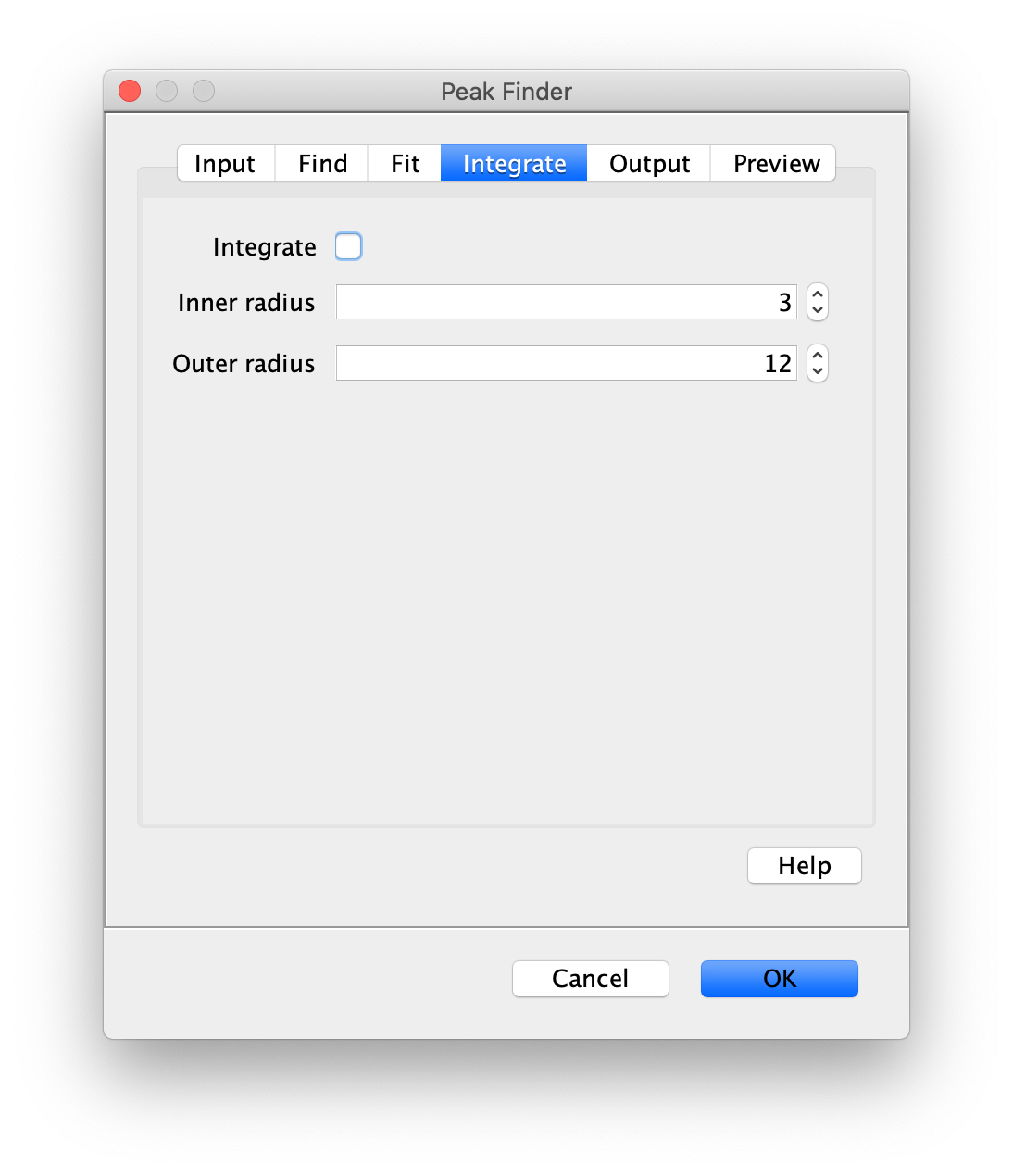
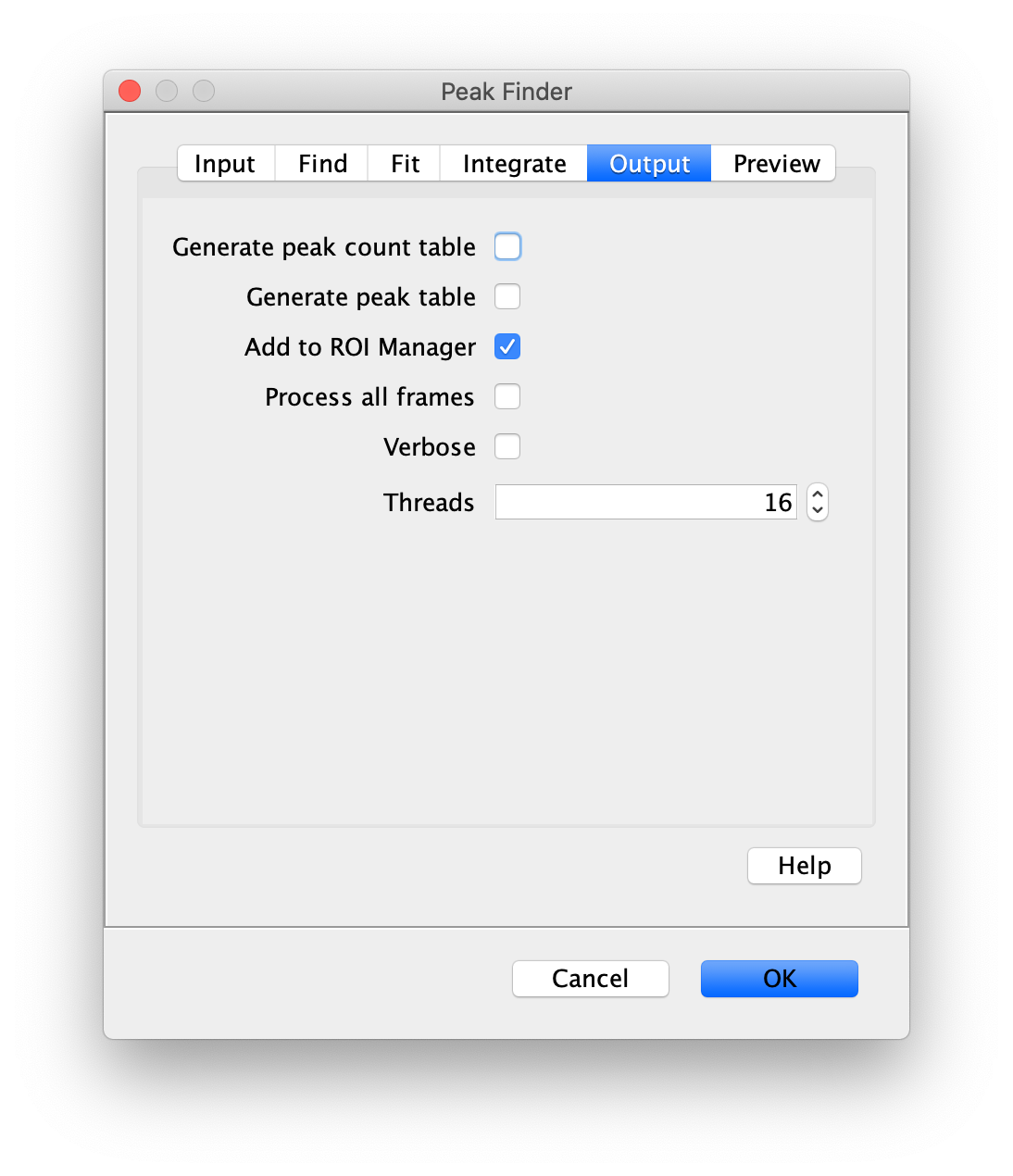
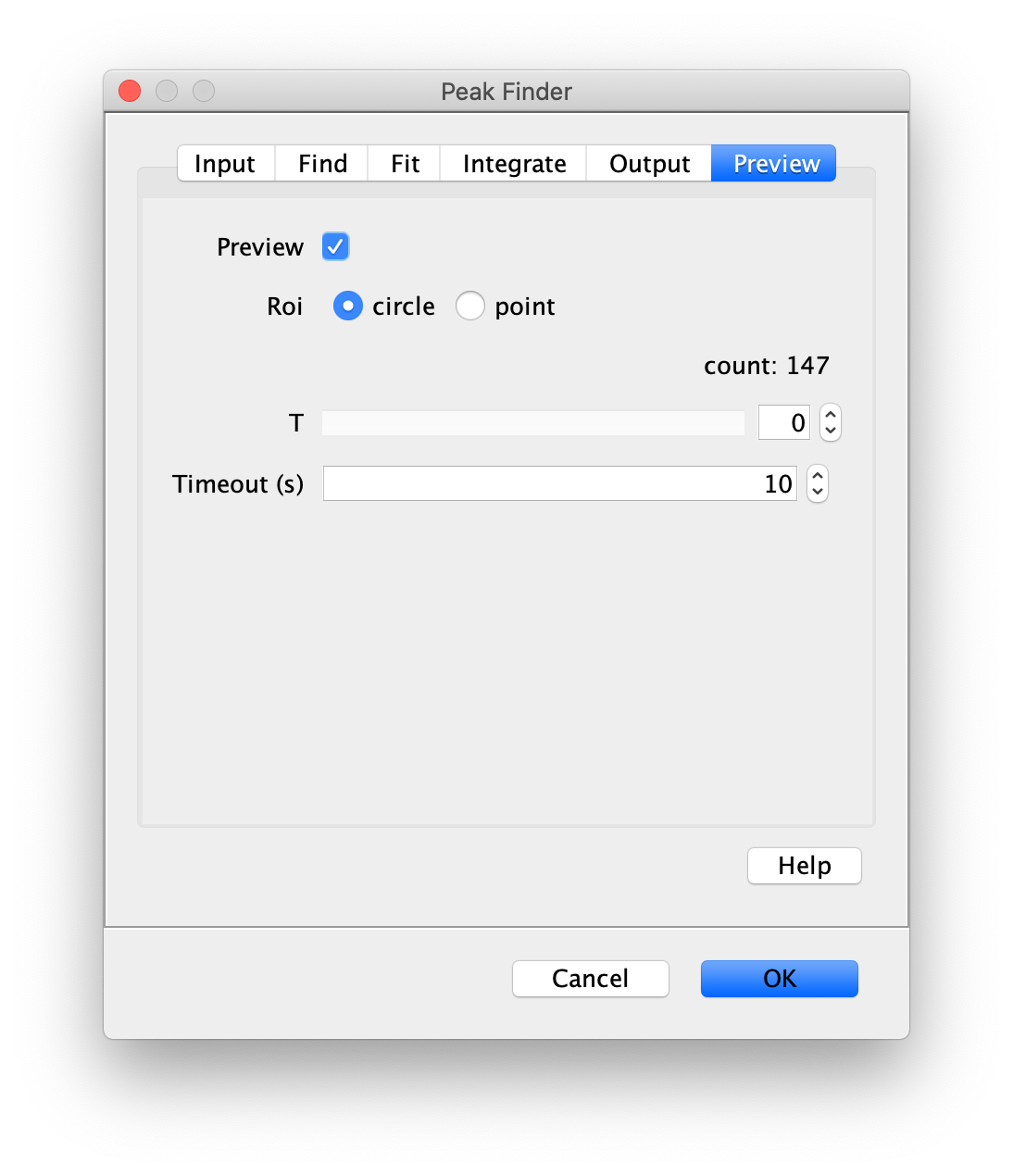
Note: We do not integrate the peaks using the Peak Finder command (The integrate checkbox is not active) because this would only provide a table with the currently selected peaks. Instead we will use the Molecule Integrator (multiview) in a later step, which produces a Molecule Archive that contains the integrated signals from all peak populations.
Transform the ROIs to the right region of the dual view
Next, transform the peak ROIs to the right region of the split view. In this way, in the next analysis step, integration of the peak intensities will be possible at both emission wavelengths at the same time. The transformation is done with the Transform ROIs command (Plugins>Mars>ROI>Transform ROIs). Use the Affine2D matrix as calculated before (see table 1). To ensure we only consider molecules with donor and acceptor signal, we will switch to the FRET channel (C=1) and apply the ‘colocalize’ filter so only peaks that are found in both red and green regions remain. Press ok to add the transformed ROIs to the ROI Manager. ROI names will have the format: UID_Red and UID_Green for emission from the same molecule using the region names, Red and Green, defined in the Output tab of the dialog.
Integrate molecules
Now switch to the full video (instead of the z projection) by clicking on its window. Then, integrate the peaks using the Molecule Integrator (multiview) command developed for multiview microscopy data (Plugins>Mars>Image>Molecule Integrator (multiview)). Enter the setting below, press ok, and a Molecule Archive with the results will open automatically upon completion of the calculation. Now the generation of the first archive, the FRET archive, is complete. Select the single metadata record in the Molecule Archive and add the FRET tag. This is required to ensure the population integrated is correctly annotated in the next steps. For this example, also add the 1-lo tag, this is used during the final analysis in the static FRET example jupyter notebook. Save the Molecule Archive with the name FRET_Archive.yama into a folder for this project.
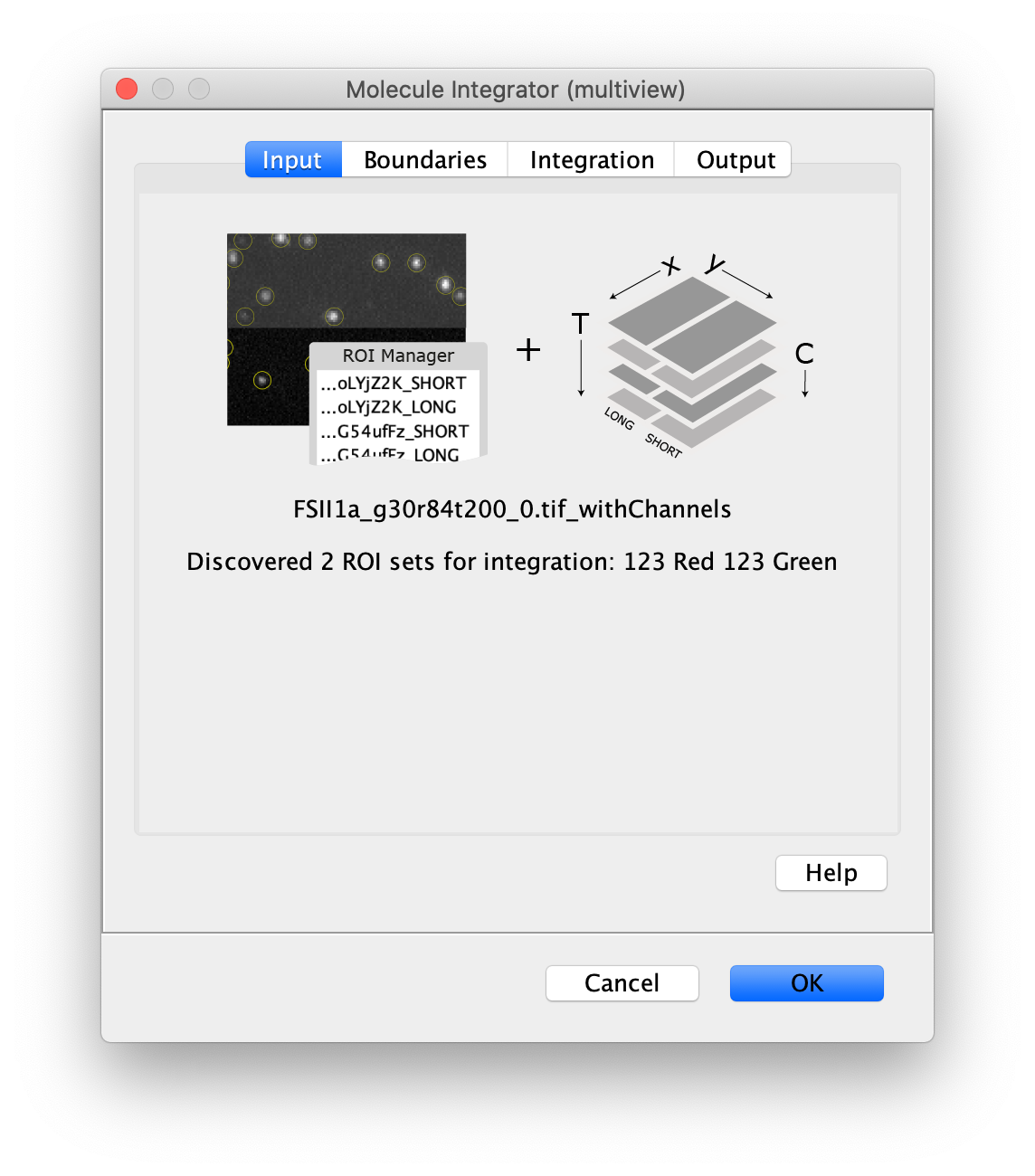
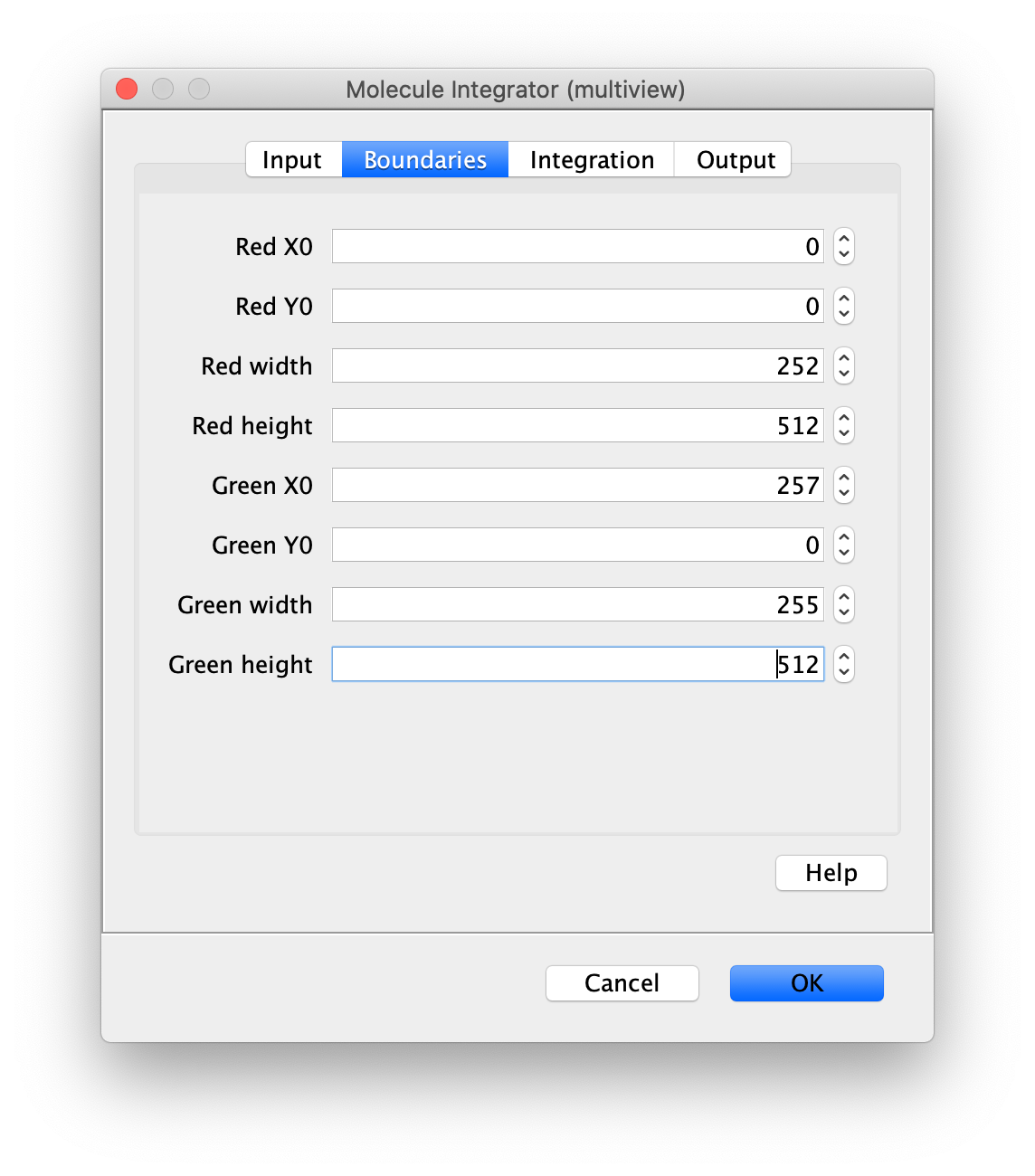
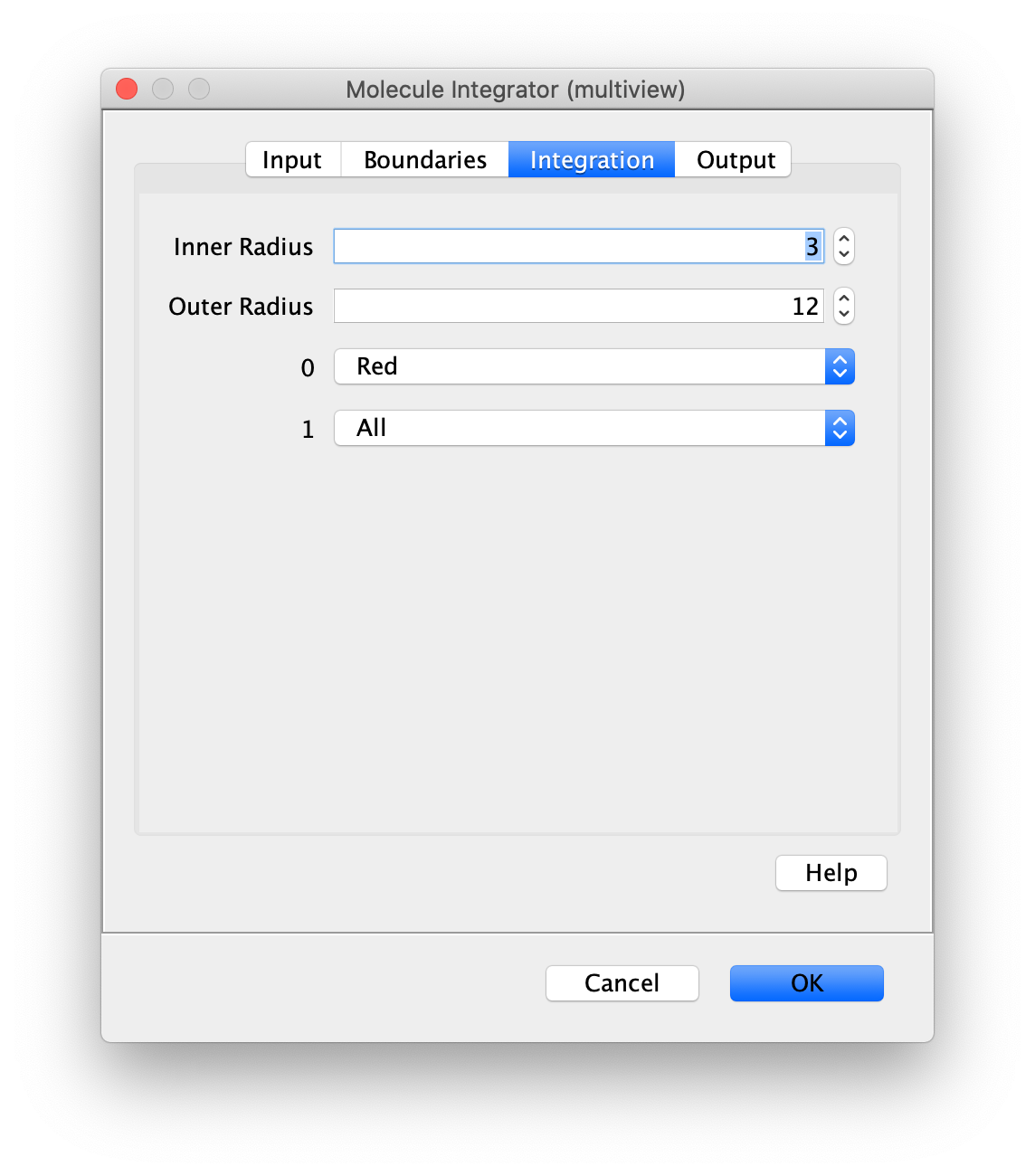
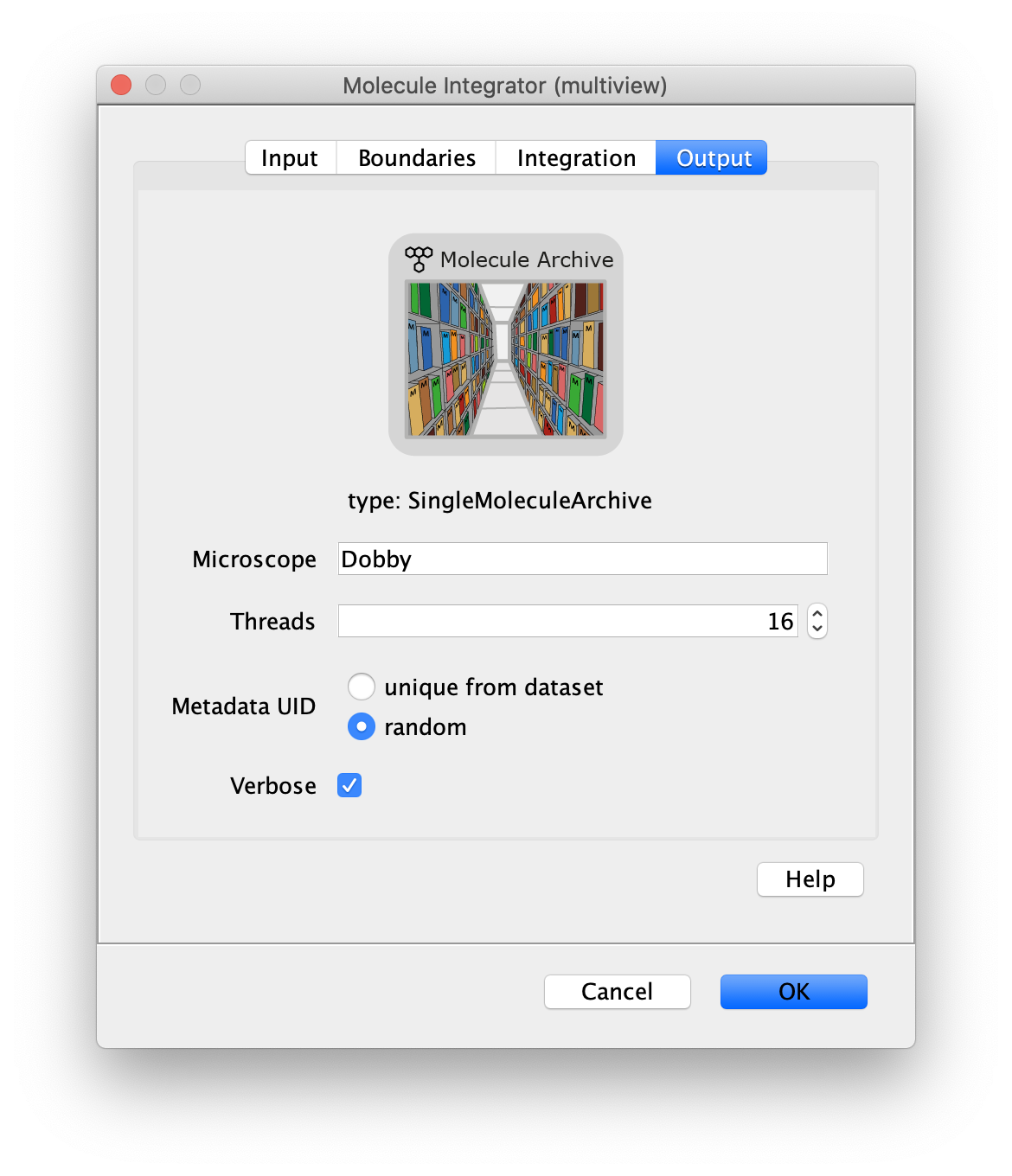
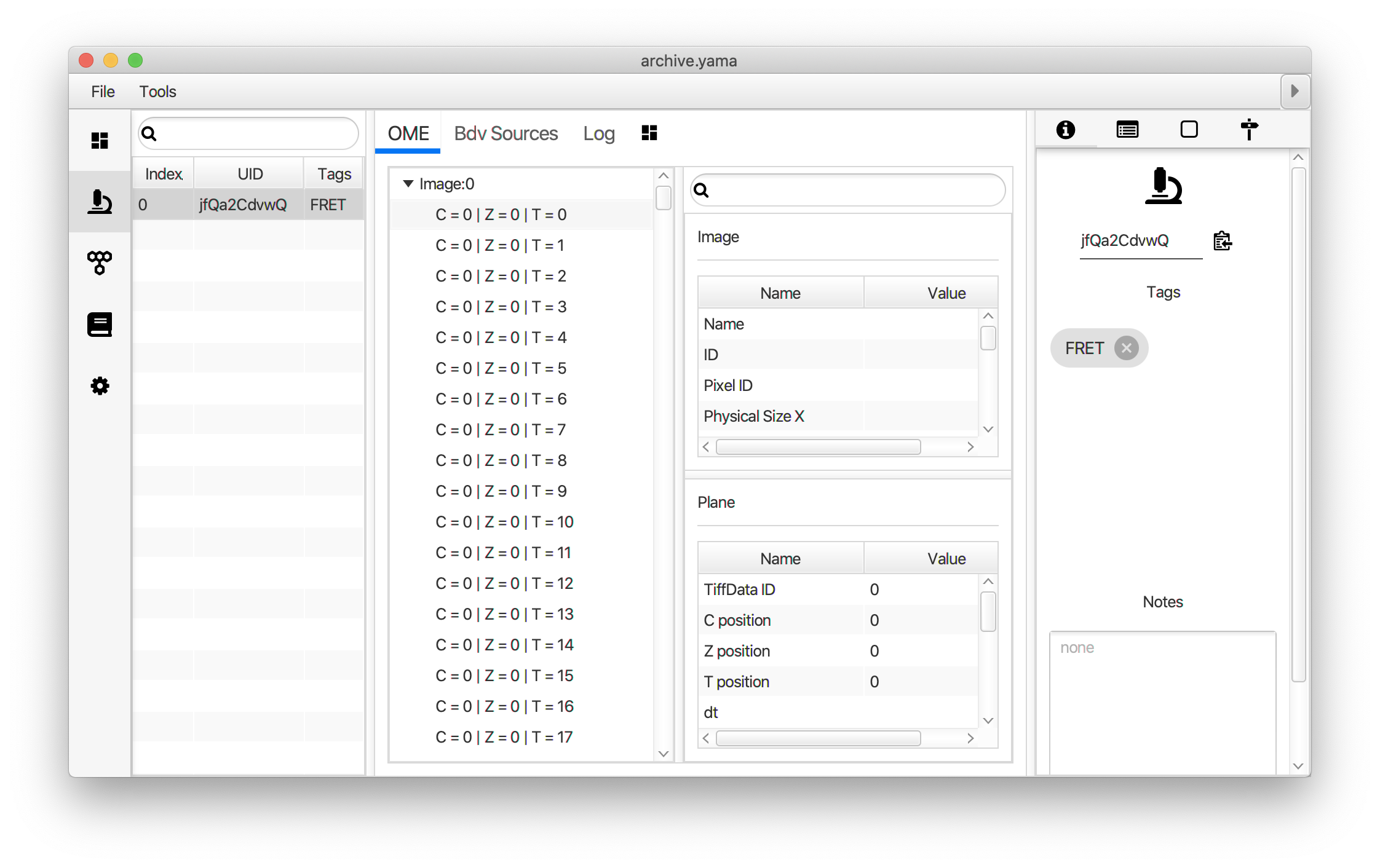
The Molecule Integrator (multiview) command names the fluorescence intensity columns found in the table of each molecule record based on the channel (excitation) and region (emission) names with the format: channel_region. The region names used are specified in the Output tab of the Transform ROIs dialog. The channel names are taken from the video metadata. For the sample data are present in the metadata, so channel indexes are used as the names (0, 1, etc.).
| Intensity parameter | Name in Mars |
|---|---|
| Iaemaex | “0_Red” (corresponding to C=0, red excitation, red emission) |
| Iaemdex | “1_Red” (corresponding to C=1, green excitation, red emission) |
| Idemdex | “1_Green” (corresponding to C=1, green excitation, green emission) |
Table 2: Overview of definitions of intensity values with their corresponding names in the Molecule Archive.
The AO Archive
First, make sure to clear all entries from the ROI Manager using the Delete button with no ROI selected. This will ensure the new collection of AO ROIs are not added to the previous set of ROIs used for FRET. To create the acceptor only (AO) Archive, follow the same procedure as outlined in section ‘The FRET Archive’ with the only difference being that in this case colocalizing peaks should be filtered. This is done by checking the ‘Remove colocalizing ROIs’ in the Transform ROIs dialog under the Colocalize tab for channel 1.
Tag the single metadata record in the resulting Molecule Archive with AO and save it with the name AO_Archive.yama. For this example, also add the 1-lo tag, this is used during the final analysis in the static FRET example jupyter notebook
The DO Archive
First, make sure to clear all entries from the ROI Manager using the Delete button with no ROI selected. This will ensure the new collection of DO ROIs are not added to the previous set of ROIs used for AO. To create the donor only (DO) Archive, follow the procedure in the section ‘The FRET Archive’ with the following changes:
- Peak Finder: select the right half of the screen (Donor emission region), and with channel 1 selected in the dialog. These steps should be done on the Z Projection image for building the ROI sets. Then the Molecule Integrator (multiview) is used on the full two channel video with all time points.
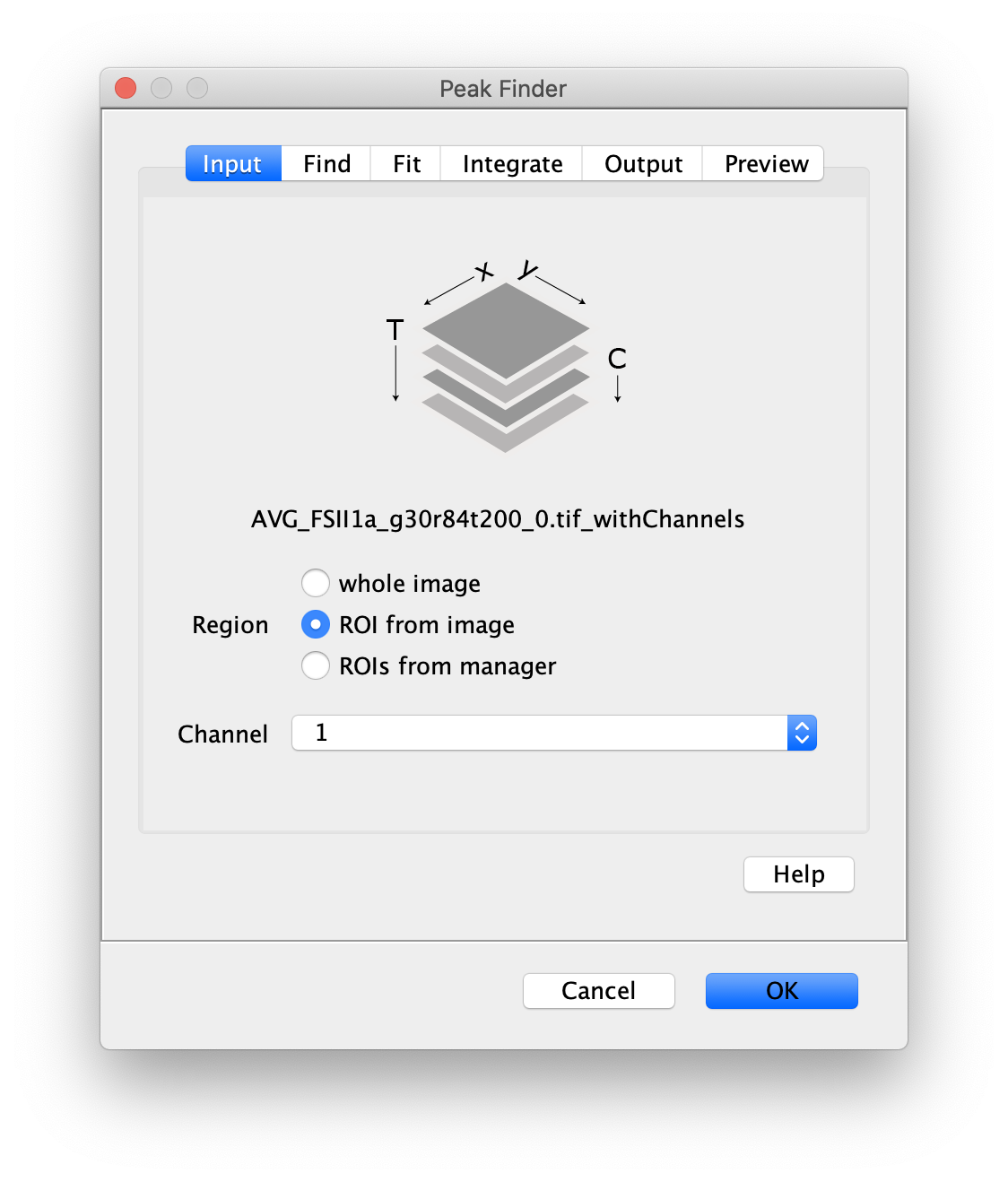
- Transform ROIs: apply the inverse transformation, set channel to 0, check ‘Remove colocalizing ROIs’ and update the region names in the Output tab to reflect a transformation from Green to Red (right to left, which is the inverse of what we used when creating the AO and FRET archives).
Tag the single metadata record in the resulting Molecule Archive with DO and save it with the name DO_Archive.yama. For this example, also add the 1-lo tag, this is used during the final analysis in the static FRET example jupyter notebook
Summary of FRET, AO and DO Archive integrations
| FRET archive | AO archive | DO archive | |
|---|---|---|---|
| Molecule | 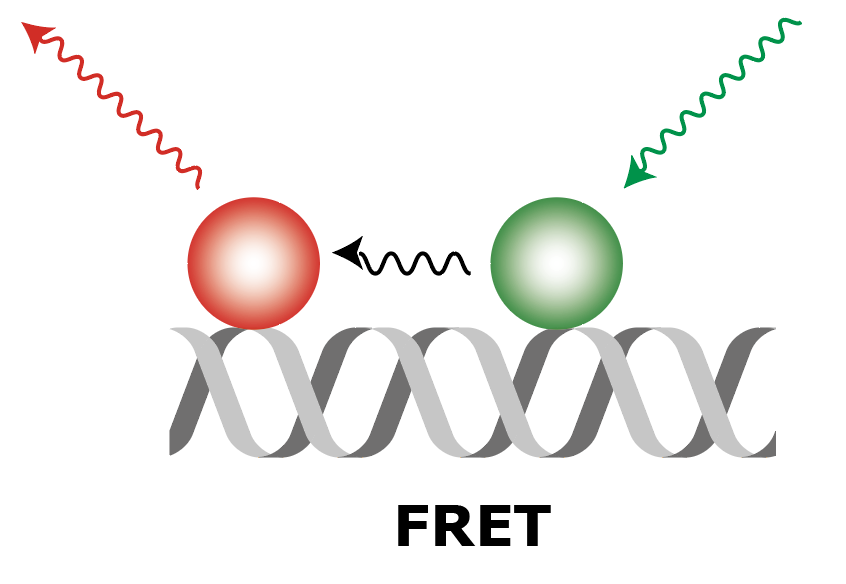 |
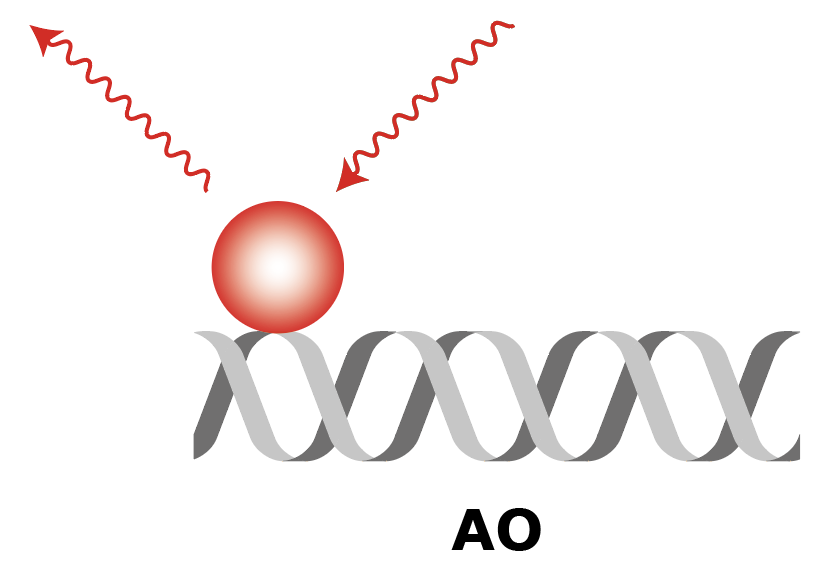 |
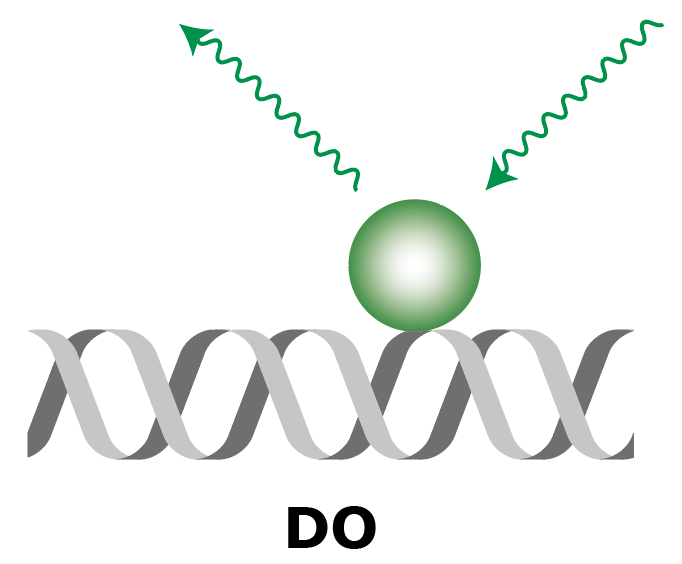 |
| What to measure | Intensity of fluorescence in red upon red excitation (Iaemaex), Intensity of fluorescence in green upon green excitation (Idemdex), Intensity of fluorescence in red upon green excitation (FRET, Iaemdex) | Intensity of fluorescence in red upon red excitation (Iaemaex), Leaking fluorescence to the green channel upon green excitation (Idemdex), Leaking fluorescence to the red channel when excited with green (Iaemdex) | Leaking fluorescence of red upon green excitation (Iaemdex), Intensity of fluorescence in green upon green excitation (Idemdex) |
| Parameter names | Iaemaex, Idemdex & Iaemdex | Iaemaex(AO), Idemdex(AO) & Iaemdex(AO) | Iaemaex(DO), Iaemdex(DO) & Idemdex(DO) |
| Analysis procedure | Find peaks in red, Transform ROIs (Affine: L->R) to colocalize (C=1) | Find peaks in red, Transform ROIs (Affine: L->R) to filter out colocalizing peaks (C=1) | Find peaks in green, Transform ROIs (Affine: R->L) to filter out colocalizing peaks (C=0) |
Merge Archives
The final step in Phase I is to merge all the archives we created into one. This is required to use the scripts in Phase II that expect AO, DO, and FRET tagged populations. Before merging the archives confirm that the metadata record in each archive is tagged with FRET, AO, or DO consistent with the steps above. If not, make sure to tag them now. All the archives (FRET_Archive.yama, AO_Archive.yama, and DO_Archive.yama) need to be placed in the same folder. Next, use the Merge Archives command (Plugins>Mars>Molecule>Merge Archive) and select the folder. When the command is finished, a merged archive is created that can be found in the same folder (merged.yama). Note: it is very important to save the archives after metadata tagging. Otherwise, the tag will not be in the merged archive and errors will be raised in the subsequent analysis steps.
Phase II
1 add molecule tags
Open the merged Molecule Archive created at the end of Phase I. This should contain three metadata records, one for each archive that was merged and they should have appropriate tags (FRET, AO or DO and all metadata records should have the 1-lo tag). Run the add molecule tags groovy script on the merged Molecule Archive. This script adds the tags on the metadata records to the corresponding molecule records. This can be confirmed by examining the tags on the records in the molecules tab. The rest of the scripts in the workflow require molecule tags.
2 profile correction
Next run the profile correction groovy script, which corrects for differences in the beam profile intensities across the field of view. This script will take the merged Molecule Archive and the beam profiles included with the sample data as inputs. You will be presented with a dialog when running the script to specify the input files that should already be open in Fiji and the column names for Aem|Aex as well as Dem|Dex. This script will add a new corrected Aem|Aex column with the suffix _Profile_Corrected that will be used in later for in the alex corrections script.

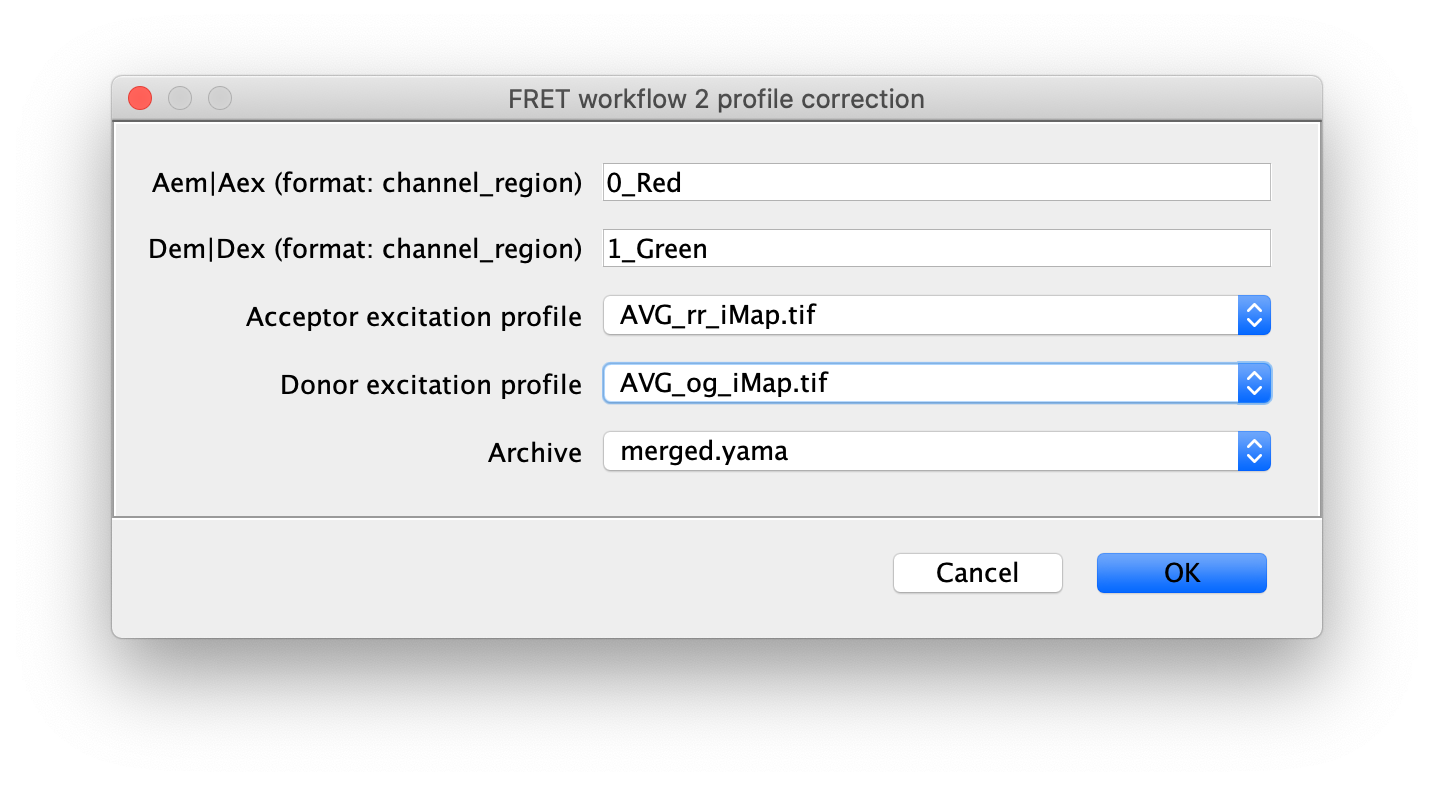
The beam profile correction images were generated using high concentrations of dye for even illumination.
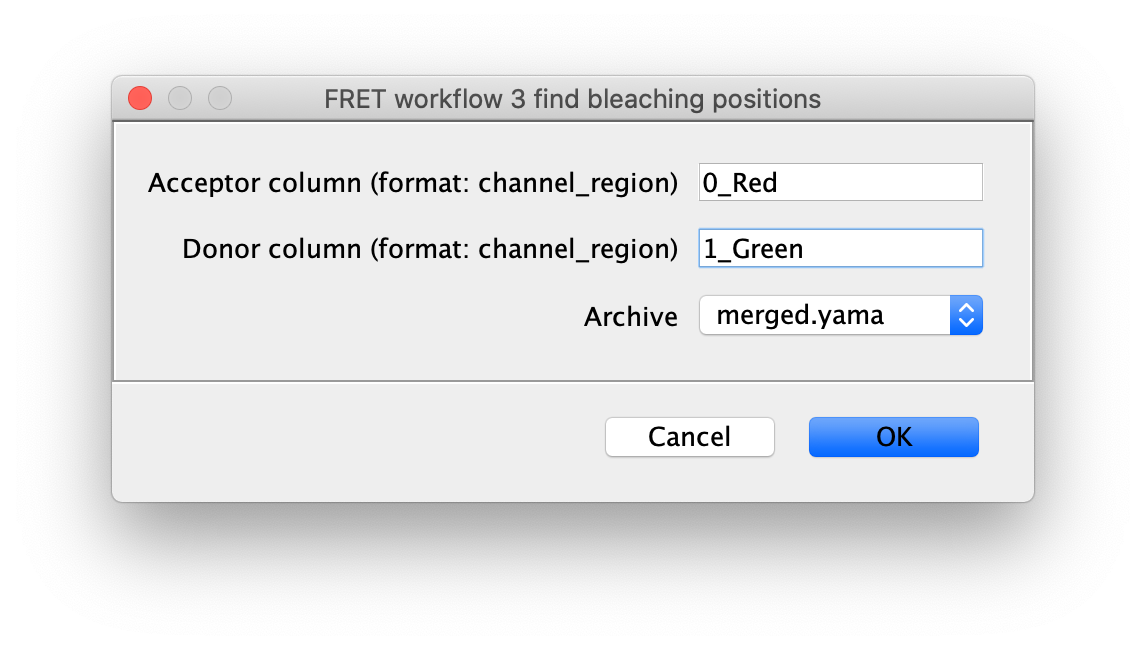
3 find bleaching positions
To correctly assess the FRET parameters, it is important to find out the bleaching time points of both dyes and to identify the first dye that bleached. Only the frames of the video up until that point are relevant for FRET. Find the bleaching positions using the find bleaching positions groovy script.
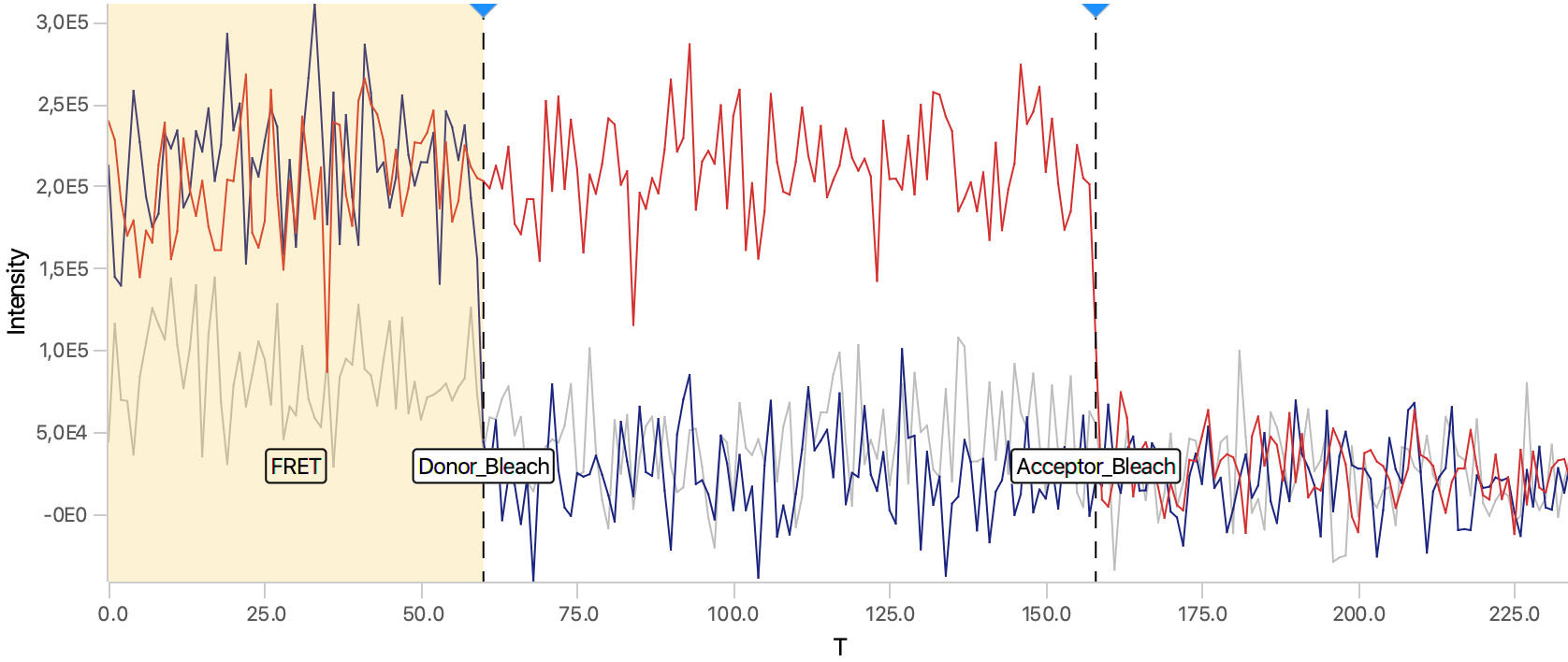
As seen below, this adds the positions Bleach_Red and Bleach_Green to molecule records. To identify the bleaching positions, the script uses the Single Change Point Finder command, which locates the single largest intensity transition within the respective intensity trace. The subsequent scripts only consider the region prior to the first bleach event, highlighted and labeled for clarity below, as the region where FRET occurred.
Plot the traces and add the Accepted tag to passing molecules
Open the plot sub-tab in the molecules tab and add three line plots representing the three measured intensities (Iaemaex, Idemdex & Iaemdex), if applicable, for each molecule. To do so select the options as shown in the screenshot. The red line represents Iaemaex (AO), the grey line Iaemdex (FRET), and the blue line Idemdex (DO). These are the three intensity vs. time traces that are required for further FRET calculations.
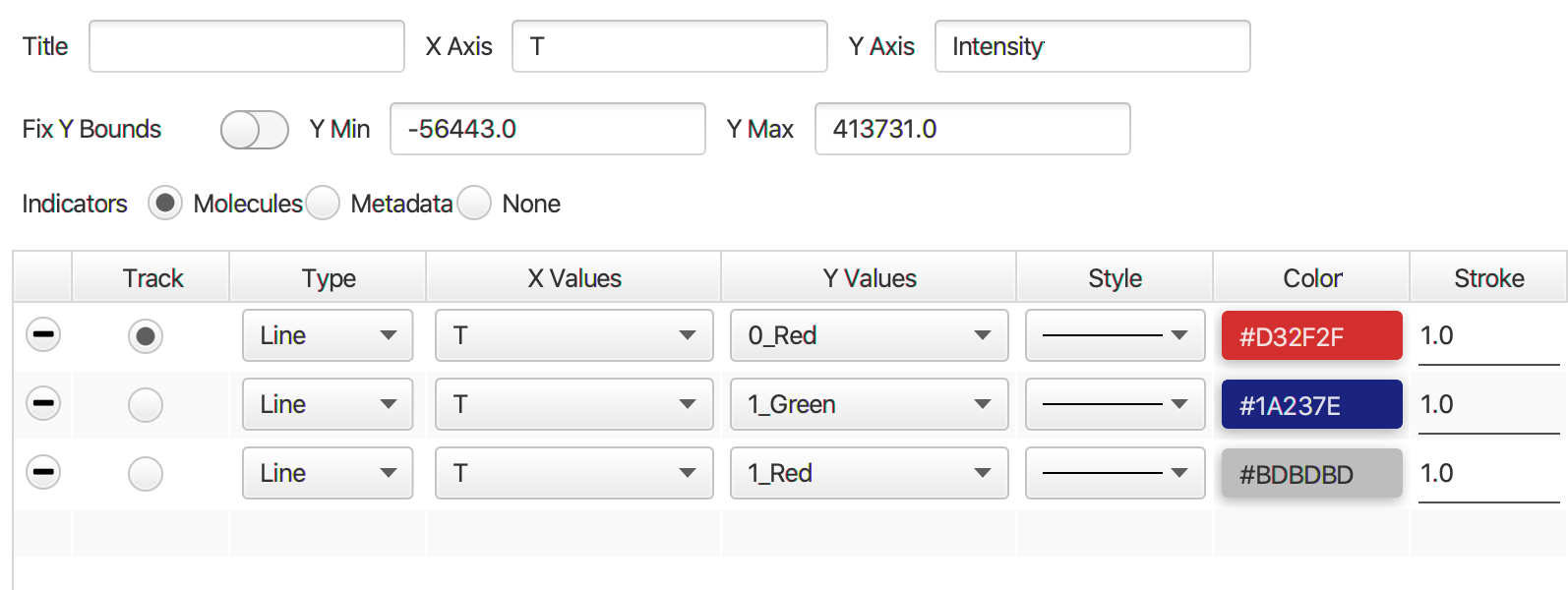
In the screenshot below the red line represents Iaemaex, the grey line Iaemdex (FRET), and the blue line Idemdex. These are the three intensity vs. time traces that are required to do further FRET calculations on.
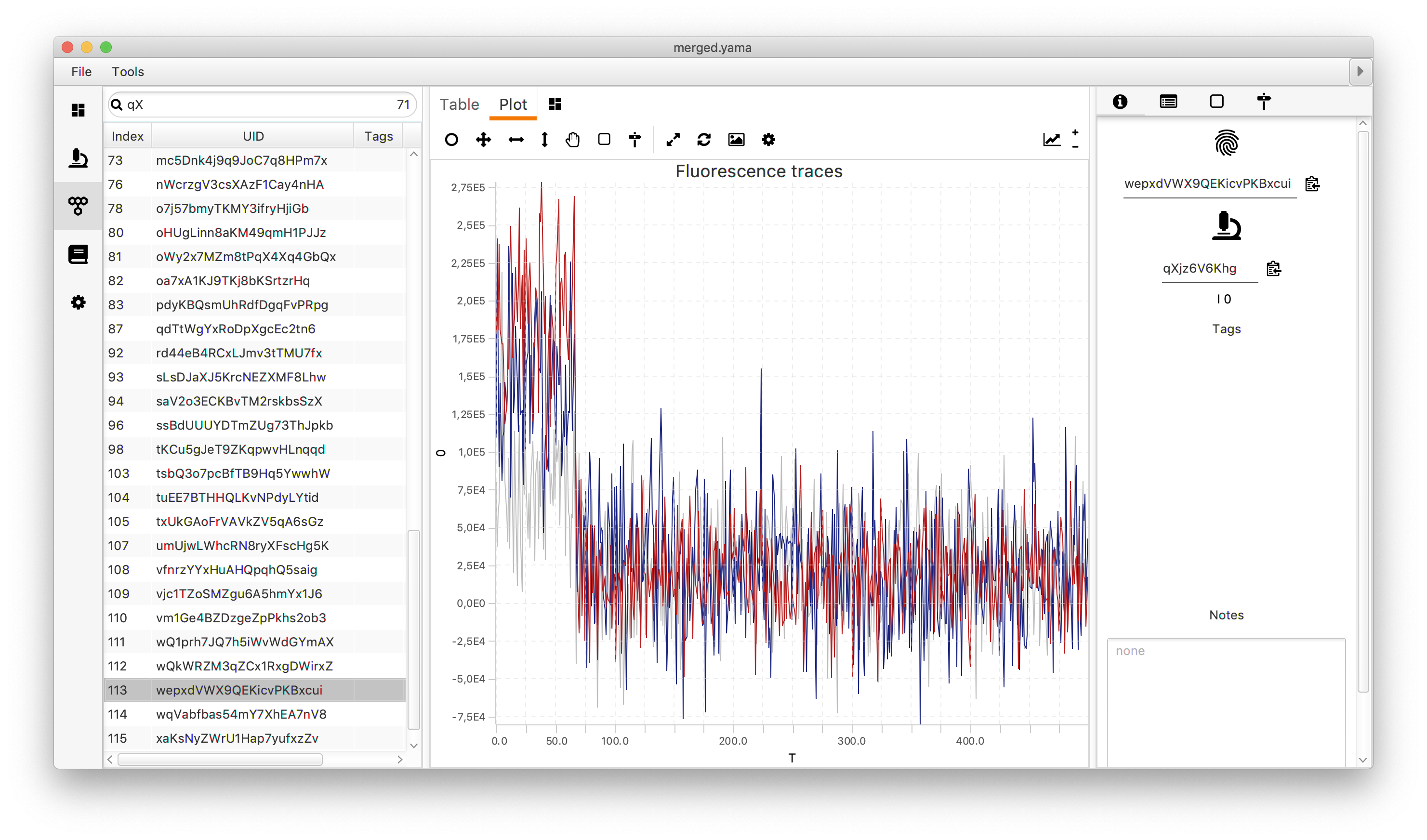
Only molecules marked with the Accepted tag will be used for further analysis. Take care to ensure a consistent set of criteria are applied when marking Accepted molecule records. We used the following criteria for this example:
- There is only 1 donor and only 1 acceptor bleaching step observed in the molecule trace
- Both donor and acceptor are present (in the case of FRET-tagged molecules)
- No major deviations or changes in signal intensity are observed that could indicate an artefact
- The fluorescent signal is significantly higher than the noise of the background
- Donor and acceptor signals are anti-correlated in the FRET region
- There is sufficient FRET life time prior to the first dye bleach event
The selection of suitable AO and DO molecules is equally important to ensure accurate determination of the alpha and delta correction factors that will significantly influence the calculation of beta and gamma. Longer lifetimes, high signal-to-noise, and low background are particularly important. The results of our classification can be directly examined in the 1-lo_1-mid_merged.yama and accompanying 1-lo_1-mid_merged.yama.rover file final archive for this example that is available from the mars-tutorials repository.
The hot key combinations for molecule tagging can be set in the settings tab of the Molecule Archive. This speeds up the process of tagging significantly. Finally, a validation notebook provided in the mars-tutorials repository offers a report with several measures for the quality of the final set Accepted molecules.
4 alex corrections
Once you have completed the analysis steps above, you are left with a Molecule Archive containing the results from the FSII1a_g30r84t200_0.tif video. This will have only a limited number of data points, which affects the accuracy of the calculated correction factors and E and S values. Moreover, reliable beta and gamma corrections are only obtained when the data consists of multiple populations (1-lo and 1-mid). Therefore, to increase the number of observations, the workflow should be repeated for all the videos and the resulting Molecule Archives should be merged together for further analysis. This should include both 1-lo and 1-mid datasets providing the populations needed for accurate beta and gamma calculations. Note that in the case of the 1-mid datasets, the 1-mid tag should be added to the metadata records instead of the 1-lo tag. This will ensure the populations can be separately analyzed in the final static FRET example jupyter notebook.
Run the alex corrections groovy script on the merged Molecule Archive containing all 1-lo and 1-mid data with tagged Accepted molecules. This script calculates all alex correction factors to generate the fully corrected I vs. T traces as well as the FRET efficiency (E) and stoichiometry (S) values. There are two options for the model used to calculate the gamma correction, which corrects for differences in excitation and detection factors between donor and acceptor. The ‘static molecules’ model should be used for this dataset. The ‘static molecules’ model assumes the FRET state of each molecule reflects a fixed constant distance, which is true for the Hellenkamp dataset. The archive used should contain merged results from the analysis of both 1-lo and 1-mid videos. The ‘static molecule’ model will use the mean E and S values from each Accepted molecule to fit the gamma correction factor. This script can be re-run multiple times if molecule tags are updated or additional data is merged in to update the corrections and final E and S values.
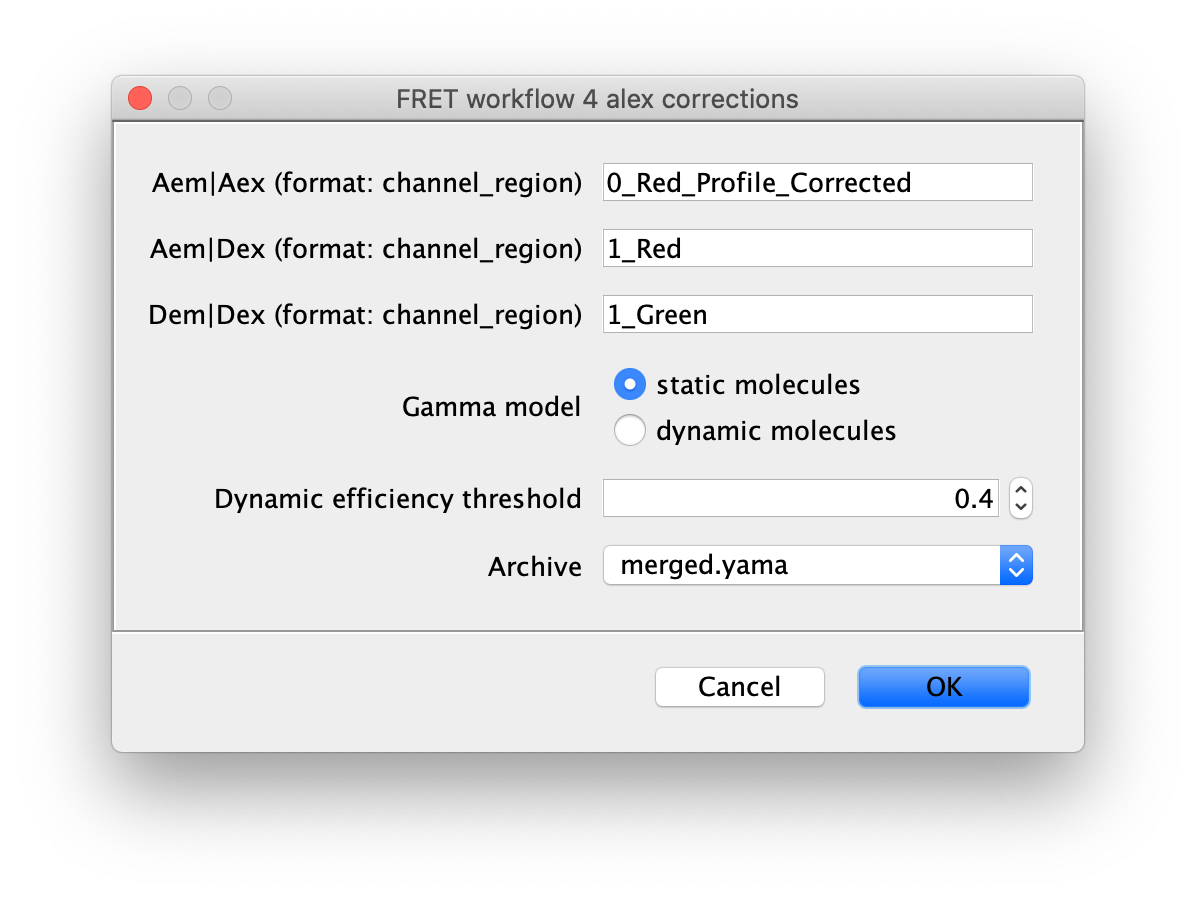
Comment: If your molecules exhibit dynamic switching between FRET states within individual traces, the ‘dynamic molecules’ Gamma model should be used with a threshold E value between the states. This is illustrated in the dynamic FRET example.
The subsections below outline the various corrections performed by the alex corrections groovy script.
Trace-wise background correction
The first correction that is applied to the dataset is a background correction. In this trace-wise process the mean fluorophore intensity after fluorophore bleaching is considered to be the background signal and is subtracted from the calculated fluorophore intensity. This is done for each intensity measurement (Iaemaex, Idemdex & Iaemdex) separately and in a trace-wise manner. The values of the corrected I parameters are stored in the archive as iiIaemaex, iiIdemdex & iiIaemdex respectively. The mean background intensities are stored as molecule parameters (1_Green_Background, 1_Red_Background, and 0_Red_Background).
Correction for leakage ($\alpha$) and direct excitation ($\delta$)
In the next step, two data corrections are carried out: a correction for leakage, the process of donor emission in the acceptor detection channel, and a correction for direct excitation, the process of acceptor emission by direct excitation of the acceptor at the donor wavelength. Both lead to signal intensity distortions when uncorrected and would influence the measured FRET parameters. Therefore they are corrected in this part of the analysis procedure. For more information about these correction parameters the reader is referred to literature9.
The leakage ($\alpha$) and direct excitation ($\delta$) correction factors can be calculated using the formulae below. As indicated, these formulae require the calculated fluorescence intensities from the DO and AO populations respectively. Subsequently, they are implemented in the latter three formulae to find the corrected E and S values. The $\alpha$ and $\delta$ correction factors as well as the calculated FA|D, iiiEapp(DO), and iiiSapp(AO) values will appear as parameters in the archive.
\[\begin{equation} \alpha = \frac{\langle ^{ii}E_{app}^{(DO)} \rangle}{1 - \langle ^{ii}E_{app}^{(DO)} \rangle} \quad\mathrm{and}\quad \delta = \frac{\langle ^{ii}S_{app}^{(AO)} \rangle}{1 - \langle ^{ii}S_{app}^{(AO)} \rangle} \end{equation}\] \[\begin{equation} F_{A|D}=^{ii}I_{Aem|Dex} - \alpha ^{ii}I_{Dem|Dex} - \delta ^{ii}I_{Aem|Aex} \end{equation}\] \[\begin{equation} ^{iii}E_{app} = \frac{F_{A|D}}{F_{A|D} + ^{ii}I_{Dem|Dex}} \quad\mathrm{and}\quad ^{iii}S_{app} = \frac{F_{A|D} + ^{ii}I_{Dem|Dex}}{F_{A|D} + ^{ii}I_{Dem|Dex} + ^{ii}I_{Aem|Aex}} \end{equation}\]Correction for excitation ($\beta$) and detection ($\gamma$) factors
The last data correction step corrects for excitation ($\beta$), normalizing excitation intensities and cross-sections of both acceptor and donor, and detection factors ($\gamma$), normalizing effective fluorescence quantum yields and detection efficiencies of both donor and acceptor. To find both global correction factors a linear regression analysis is performed with the following formula. Note: Each value of iiiEapp and iiiSapp is included in the regression. This means that longer traces have a larger weight in the final fit since these traces contribute a larger number of data points to the dataset used to make a fit.
\[\begin{equation} ^{iii}S_{app}^{(FRET)} = \frac{1}{1 + \gamma * \beta + (1-\gamma)*\beta *^{iii}E_{app}^{(FRET)}} \end{equation}\] \[\begin{equation} \frac{1}{^{iii}S_{app}^{(FRET)}} = 1 + \gamma* \beta + (1-\gamma)*\beta *^{iii}E_{app}^{(FRET)} = b * ^{iii}E_{app}^{(FRET)} + a \end{equation}\]From this equation the dependencies of $\beta$ and $\gamma$ on a and b can be deducted to:
\[\begin{equation} \beta = a + b - 1 \quad\mathrm{and}\quad \gamma = \frac{a - 1}{a + b - 1} \end{equation}\]Once the values of $\beta$ and $\gamma$ have been calculated, the fully corrected E and S values can be calculated. To do so, first FD|D and FA|A are calculated, then added to the archive, followed by the calculation of E and S. Note that these values are extremely sensitive to outliers and will have low significance when used on datasets with a low number of data points.
\[\begin{equation} F_{D|D} = \gamma * ^{ii}I_{Dem|Dex} \quad\mathrm{and}\quad F_{A|A} = \frac{1}{\beta} * ^{ii}I_{Aem|Aex} \end{equation}\] \[\begin{equation} E = \frac{F_{A|D}}{F_{D|D} + F_{A|D}} \quad\mathrm{and}\quad S = \frac{F_{A|D} + F_{D|D}}{F_{D|D} + F_{A|D} + F_{A|A}} \end{equation}\]Molecule table columns
| Molecule table column | Description | Channel | Region | Added by |
|---|---|---|---|---|
| T | OME time points | all | all | Molecule Integrator (multiview) |
| 0_Red_Time_(s) | Time in seconds if discovered in the image metadata | 0 | Red | Molecule Integrator (multiview) |
| 0_Red_X | X coordinate of molecule position | 0 | Red | Molecule Integrator (multiview) |
| 0_Red_Y | Y coordinate of molecule position | 0 | Red | Molecule Integrator (multiview) |
| 0_Red | Local median background corrected integrated intensity | 0 | Red | Molecule Integrator (multiview) |
| 0_Red_Median_Background | Local median background intensity | 0 | Red | Molecule Integrator (multiview) |
| 0_Red_Uncorrected | Raw integrated intensity with no background correction | 0 | Red | Molecule Integrator (multiview) Verbose mode |
| 0_Red_Mean_Background | Local mean background intensity | 0 | Red | Molecule Integrator (multiview) Verbose mode |
| 1_Red_Time_(s) | Time in seconds if discovered in the image metadata | 0 | Red | Molecule Integrator (multiview) |
| 1_Red_X | X coordinate of molecule position | 1 | Red | Molecule Integrator (multiview) |
| 1_Red_Y | Y coordinate of molecule position | 1 | Red | Molecule Integrator (multiview) |
| 1_Red | Local median background corrected integrated intensity | 1 | Red | Molecule Integrator (multiview) |
| 1_Red_Median_Background | Local median background intensity | 1 | Red | Molecule Integrator (multiview) |
| 1_Red_Uncorrected | Raw integrated intensity with no background correction | 1 | Red | Molecule Integrator (multiview) Verbose mode |
| 1_Red_Mean_Background | Local mean background intensity | 1 | Red | Molecule Integrator (multiview) Verbose mode |
| 1_Green_Time_(s) | Time in seconds if discovered in the image metadata | 1 | Green | Molecule Integrator (multiview) |
| 1_Green_X | X coordinate of molecule position | 1 | Green | Molecule Integrator (multiview) |
| 1_Green_Y | Y coordinate of molecule position | 1 | Green | Molecule Integrator (multiview) |
| 1_Green | Local median background corrected integrated intensity | 1 | Green | Molecule Integrator (multiview) |
| 1_Green_Median_Background | Local median background intensity | 1 | Green | Molecule Integrator (multiview) |
| 1_Green_Uncorrected | Raw integrated intensity with no background correction | 1 | Green | Molecule Integrator (multiview) Verbose mode |
| 1_Green_Mean_Background | Local mean background intensity | 1 | Green | Molecule Integrator (multiview) Verbose mode |
| 0_Red_Profile_Corrected | Local median background corrected, beam profile corrected, integrated intensity | 0 | Red | 2 profile correction |
| iiIdemdex | Donor emission after subtracting mean background after photobleaching | 1 | Green | 4 alex corrections |
| iiIaemaex | Acceptor emission after subtracting mean background after photobleaching | 0 | Red | 4 alex corrections |
| iiIaemdex | Acceptor emission from FRET after subtracting mean background after photobleaching | 1 | Red | 4 alex corrections |
| iiEapp | FRET efficiency after correcting for background after photobleaching | - | - | 4 alex corrections |
| iiSapp | Stoichiometry after correcting for background after photobleaching | - | - | 4 alex corrections |
| FAD | Acceptor emission from FRET corrected for background, leakage ($\alpha$), direct excitation ($\delta$) | 1 | Red | 4 alex corrections |
| iiiEapp | FRET efficiency after correcting for background, leakage ($\alpha$), direct excitation ($\delta$) | - | - | 4 alex corrections |
| iiiSapp | Stoichiometry after correcting for background, leakage ($\alpha$), direct excitation ($\delta$) | - | - | 4 alex corrections |
| FDD | Donor emission after correcting for background, excitation ($\beta$) and detection ($\gamma$) factors | 1 | Green | 4 alex corrections |
| FAA | Acceptor emission after correcting for background, excitation ($\beta$) and detection ($\gamma$) factors | 1 | Green | 4 alex corrections |
| SUM_Dex | Sum of acceptor and donor emission during FRET (FAD + FDD). Expected to be stable in FRET region. | - | - | 4 alex corrections |
| SUM_signal | Sum of all dye emissions (FAA + FAD + FDD). Expected to be stable in FRET region. | - | - | 4 alex corrections |
| E | Fully corrected FRET efficiency (background, $\alpha$, $\delta$, $\beta$, $\gamma$) | - | - | 4 alex corrections |
| S | Fully corrected stoichiometry (background, $\alpha$, $\delta$, $\beta$, $\gamma$) | - | - | 4 alex corrections |
Molecule parameters
The following molecule parameters provide mean values for calculated FRET properties and validation measures. They are added by the alex corrections groovy script.
| Molecule parameter | Description |
|---|---|
| iiEapp | Mean FRET efficiency after correcting for background after photobleaching |
| iiSapp | Mean Stoichiometry after correcting for background after photobleaching |
| iiiEapp | Mean FRET efficiency after correcting for background, leakage ($\alpha$), direct excitation ($\delta$) |
| iiiSapp | Mean stoichiometry after correcting for background, leakage ($\alpha$), direct excitation ($\delta$) |
| E | Mean fully corrected FRET efficiency (background, $\alpha$, $\delta$, $\beta$, $\gamma$) |
| S | Mean fully corrected stoichiometry (background, $\alpha$, $\delta$, $\beta$, $\gamma$) |
| SUM_Dex_FRET_Coefficient_of_Variation | The coefficient of variation for the sum of the donor and acceptor emission in the FRET region |
| SUM_FDD_Recovery_Coefficient_of_Variation | The coefficient of variation for the sum of the donor and acceptor emission in the recovery region after acceptor bleach |
| S_Between_Bleaches | The mean value of S between dye bleach events |
| E_Between_Bleaches | The mean value of E between dye bleach events |
| SUM_signal_FRET_Coefficient_of_Variation | The coefficient of variation for the sum of the donor and acceptor emission and acceptor emission on acceptor excitation in the FRET region |
| FRET_Pearsons_Correlation | The Pearson correlation coefficient in the FRET region for donor and acceptor emission. |
| FAA_Coefficient_of_Variation | The coefficient of variation for the acceptor emission on acceptor excitation. |
Exploration in Python and Comparisons to Literature
The final Molecule Archive containing all merged data is available in the mars-tutorials repository in the files 1-lo_1-mid_merged.yama and accompanying 1-lo_1-mid_merged.yama.rover.
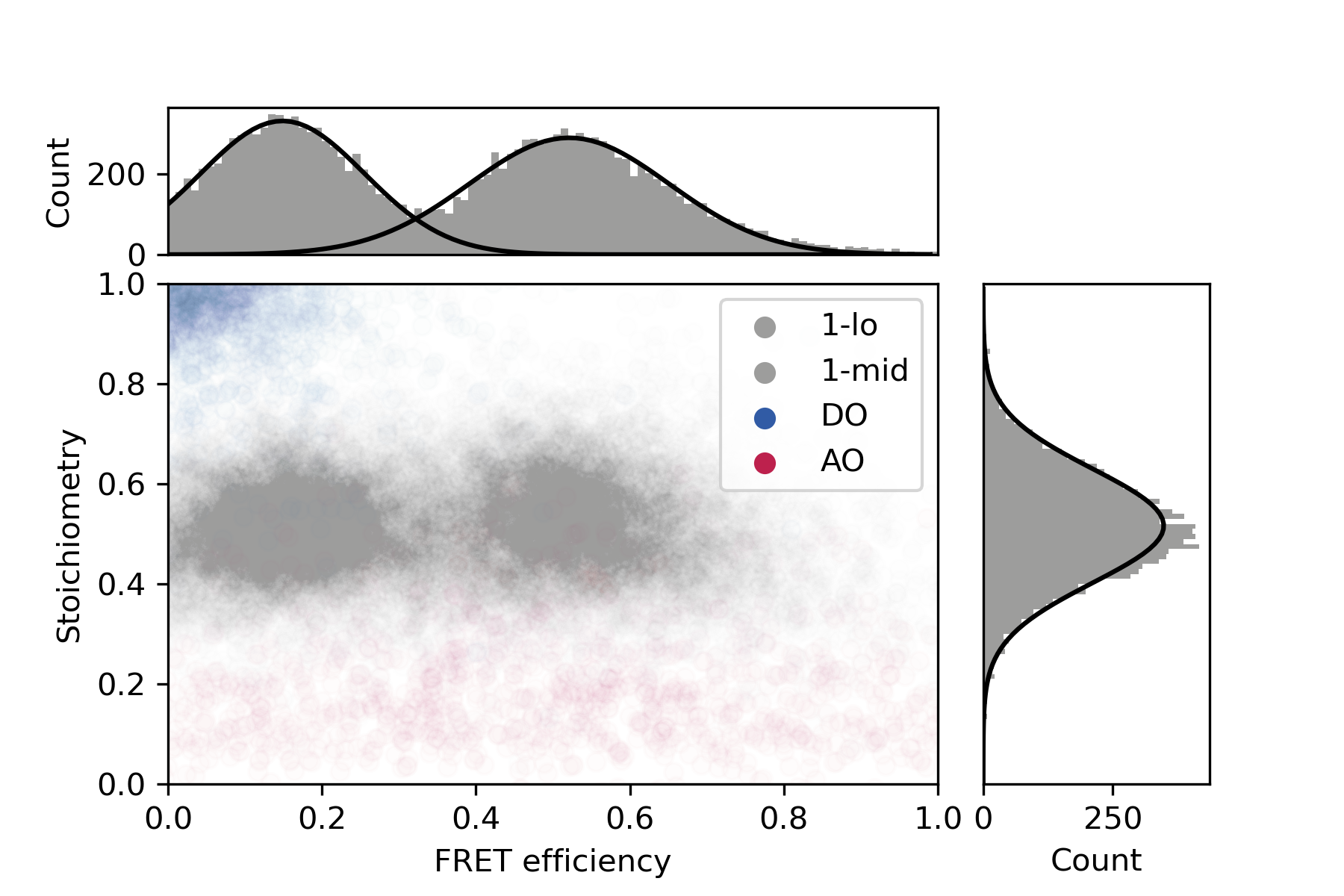
Final distributions are generated using the static FRET example jupyter notebook provided in the mars-tutorials repository. This notebook requires the file path to a local copy of 1-lo_1-mid_merged.yama and Fiji as inputs. Please note that running the notebook requires the set-up of a new python environment. Directions to do so can be found in this tutorial. After running all cells several charts are generated showing E and S values for each stage of corrections and the fully corrected distributions.
The final plot of this analysis shows that the FRET populations are observed at E= 0.15 (1-lo) and E= 0.52 (1-mid). This is in agreement with the values as published by Hellenkamp et al. 9 who reported E-values of E = 0.15 +/- 0.02 (1-lo) and E = 0.56 +/- 0.03 (1-mid) respectively.
Troubleshooting
This section highlights some of the common errors or problems that may occur during following along with the example as well as possible solutions. Please reference the table below in case you encounter any problems during the analysis. If your question has not been answered in this section, please feel free to reach out to our lab by making a forum post.
| Problem Description | Solution |
|---|---|
| Most parameters (E, S, iEapp, iSapp) display NaN values | This most often happens because some of the metadata tags are missing. Solution: check in the metadata tab if the metadata records are tagged ‘FRET’, ‘AO’, and ‘DO’ respectively. Solution: add the correct tag to the metadata. This problem often arises when the archive has been tagged, but has not been saved, before merging using the Merge Archives command. |
| No peaks were found by the Peak Finder command | This might be the case when the Peak Finder settings (especially threshold settings) are not correct. Solution: verify that the correct Peak Finder settings were entered in the dialog and run the analysis again. |
| The ROI manager is empty after running the Transform ROIs command | This behavior can be caused by two issues: (1) there were no peaks placed in the ROI Manager by the Peak Finder or (2) the ROI Manager was not closed or reset before doing running the transform ROIs command on a second dataset. Solution: (1) check if the Peak Finder settings are entered correctly and use the ‘preview’ option to verify that the command finds the peaks. Check if the ‘Add to ROI Manager’ option is selected in the tab. (2) close the ROI Manager and run the analysis again, starting from the Peak Finder command. |
| The archive or script editor freezes during analysis | We have observed this during develop on rare occasions, especially when the same Fiji session has been active on your computer for a long time. Solution: close Fiji and retry the analysis after a software restart. Saving several copies of the archive during processing is best practive. |
| No I vs. T plots are observed after entering the x and y settings in the plotting menu | Solution: refresh the plot by pressing the refresh button at the bottom right corner of the plot settings menu |
| No output is generated after running a Mars tool | Either the input settings were selected in such a way that no output is expected, or the tool encountered an error. Solution: (1) verify the settings of the tool. (2) Check for red printed errors in the Fiji Console window (Window>Console). Either resolve the error by selecting different settings in the Mars tool dialog window or run the tool again. |
| No changes are made to the Archive after running one of the scripts | It might be that the script encountered an error and did not run till completion. Solution: open the ‘Console’ window (Window>Console) in Fiji and see if any red errors are raised. If so, resolve the error and run the script again. |
References
(1) Roy, R.; Hohng, S.; Ha, T. A Practical Guide to Single-Molecule FRET. Nat. Methods 2008, 5 (6), 507–516. https://doi.org/10.1038/nmeth.1208.
(2) Stryer, L.; Haugland, R. P. Energy Transfer: A Spectroscopic Ruler. Proc. Natl. Acad. Sci. U. S. A. 1967, 58 (2), 719–726. https://doi.org/10.1073/pnas.58.2.719.
(3) Ha, T.; Enderle, T.; Ogletree, D. F.; Chemla, D. S.; Selvin, P. R.; Weiss, S. Probing the Interaction between Two Single Molecules: Fluorescence Resonance Energy Transfer between a Single Donor and a Single Acceptor. Proc. Natl. Acad. Sci. U. S. A. 1996, 93 (13), 6264–6268. https://doi.org/10.1073/pnas.93.13.6264.
(4) Mekler, V.; Kortkhonjia, E.; Mukhopadhyay, J.; Knight, J.; Revyakin, A.; Kapanidis, A. N.; Niu, W.; Ebright, Y. W.; Levy, R.; Ebright, R. H. Structural Organization of Bacterial RNA Polymerase Holoenzyme and the RNA Polymerase-Promoter Open Complex. Cell 2002, 108 (5), 599–614. https://doi.org/10.1016/s0092-8674(02)00667-0.
(5) Choi, U. B.; Strop, P.; Vrljic, M.; Chu, S.; Brunger, A. T.; Weninger, K. R. Single-Molecule FRET–Derived Model of the Synaptotagmin 1–SNARE Fusion Complex. Nat. Struct. Mol. Biol. 2010, 17 (3), 318–324. https://doi.org/10.1038/nsmb.1763.
(6) Hellenkamp, B.; Wortmann, P.; Kandzia, F.; Zacharias, M.; Hugel, T. Multidomain Structure and Correlated Dynamics Determined by Self-Consistent FRET Networks. Nat. Methods 2017, 14 (2), 174–180. https://doi.org/10.1038/nmeth.4081.
(7) Schuler, B.; Eaton, W. A. Protein Folding Studied by Single-Molecule FRET. Curr. Opin. Struct. Biol. 2008, 18 (1), 16–26. https://doi.org/10.1016/J.SBI.2007.12.003.
(8) Colombo, S.; Broggi, S.; Collini, M.; D’Alfonso, L.; Chirico, G.; Martegani, E. Detection of CAMP and of PKA Activity in Saccharomyces Cerevisiae Single Cells Using Fluorescence Resonance Energy Transfer (FRET) Probes. Biochem. Biophys. Res. Commun. 2017, 487 (3), 594–599. https://doi.org/10.1016/J.BBRC.2017.04.097.
(9) Hellenkamp, B.; Schmid, S.; Doroshenko, O.; Opanasyuk, O.; Kühnemuth, R.; Rezaei Adariani, S.; Ambrose, B.; Aznauryan, M.; Barth, A.; Birkedal, V.; et al. Precision and Accuracy of Single-Molecule FRET Measurements—a Multi-Laboratory Benchmark Study. Nat. Methods 2018, 15 (9), 669–676. https://doi.org/10.1038/s41592-018-0085-0.