Introduction to Groovy Scripting Tutorial
level: intermediate, duration: 20 min
This tutorial will introduce the basics of Mars scripting. Scripting gives the opportunity of (partly) automating data analysis. In this first tutorial the key aspects of scripting will be discussed: how to open the Fiji script editor, how to execute a script, access data from the Molecule Archive from within a script, examples for simple calculations, and coding in single lines: streams. The advanced scripting tutorial will go into more detail on less straight-forward Groovy scripting in Mars. For more information on the Groovy scripting language the reader is referred to the Groovy documentation.
These calculations can also be done in a Jupyter notebook with Python. This notebook is provided in the mars tutorials repository.
1. How to Open the Script Editor in Fiji
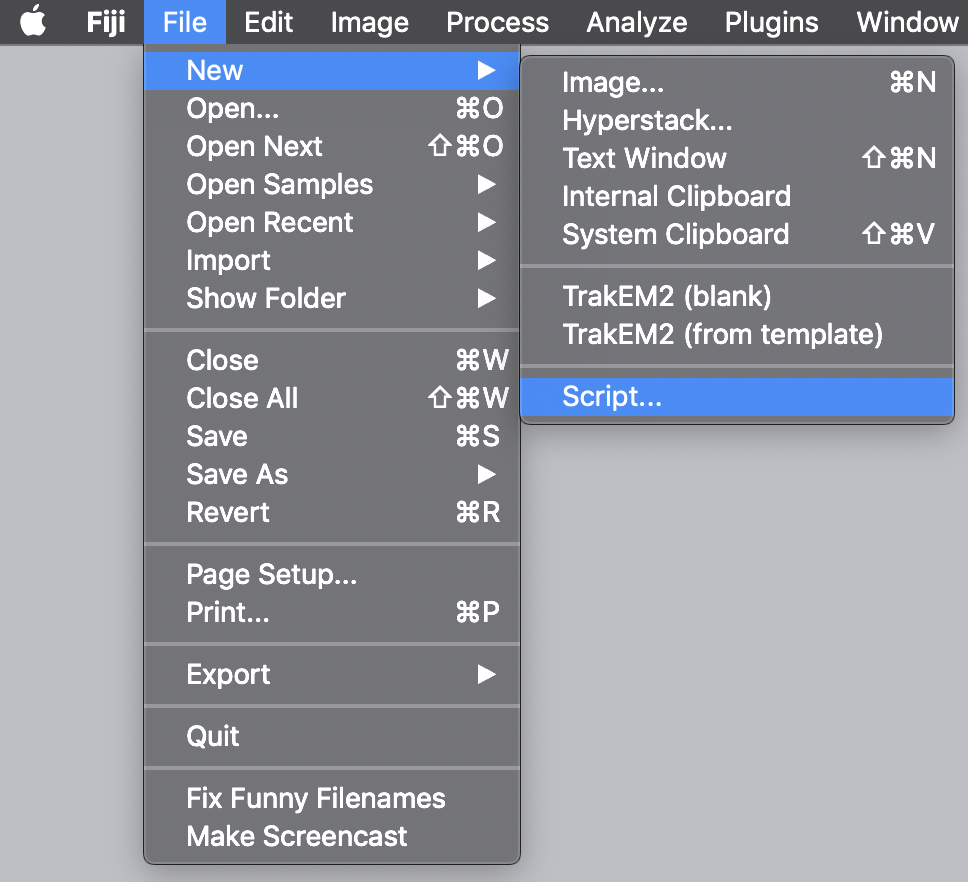
To follow along with this tutorial open the ‘TestVideo_archive_var.yama’ archive in Mars that was created in the Let’s Calculate the Variance tutorial. Alternatively, download this archive from the mars tutorials repository. To start, to open the script editor press File > New > Script in the Fiji menu as displayed in the image. This will open a new window to be used for scripting. Select the language of choice using the Language menu in the Fiji toolbar. In this case, set the language to ‘Groovy’.
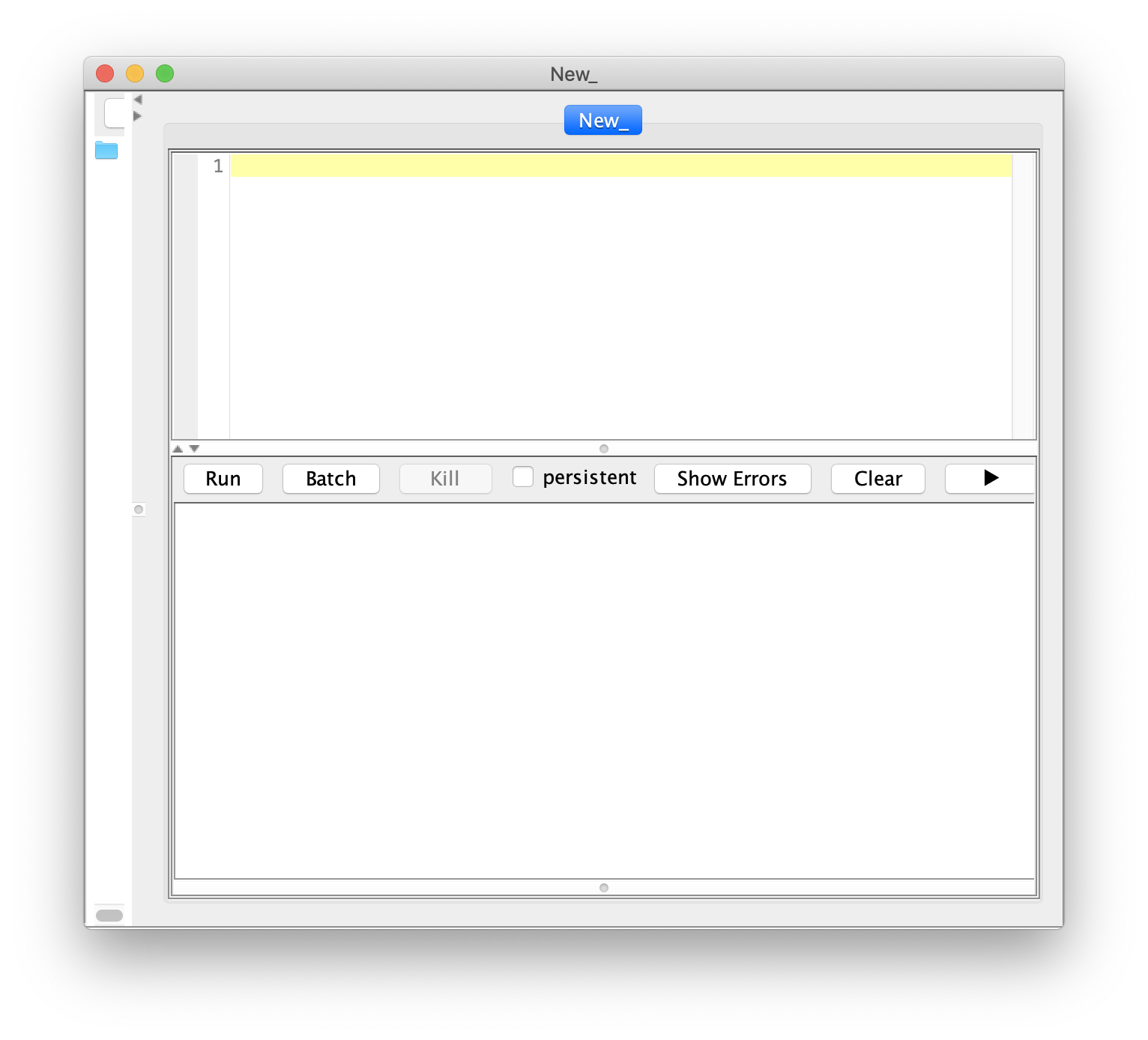
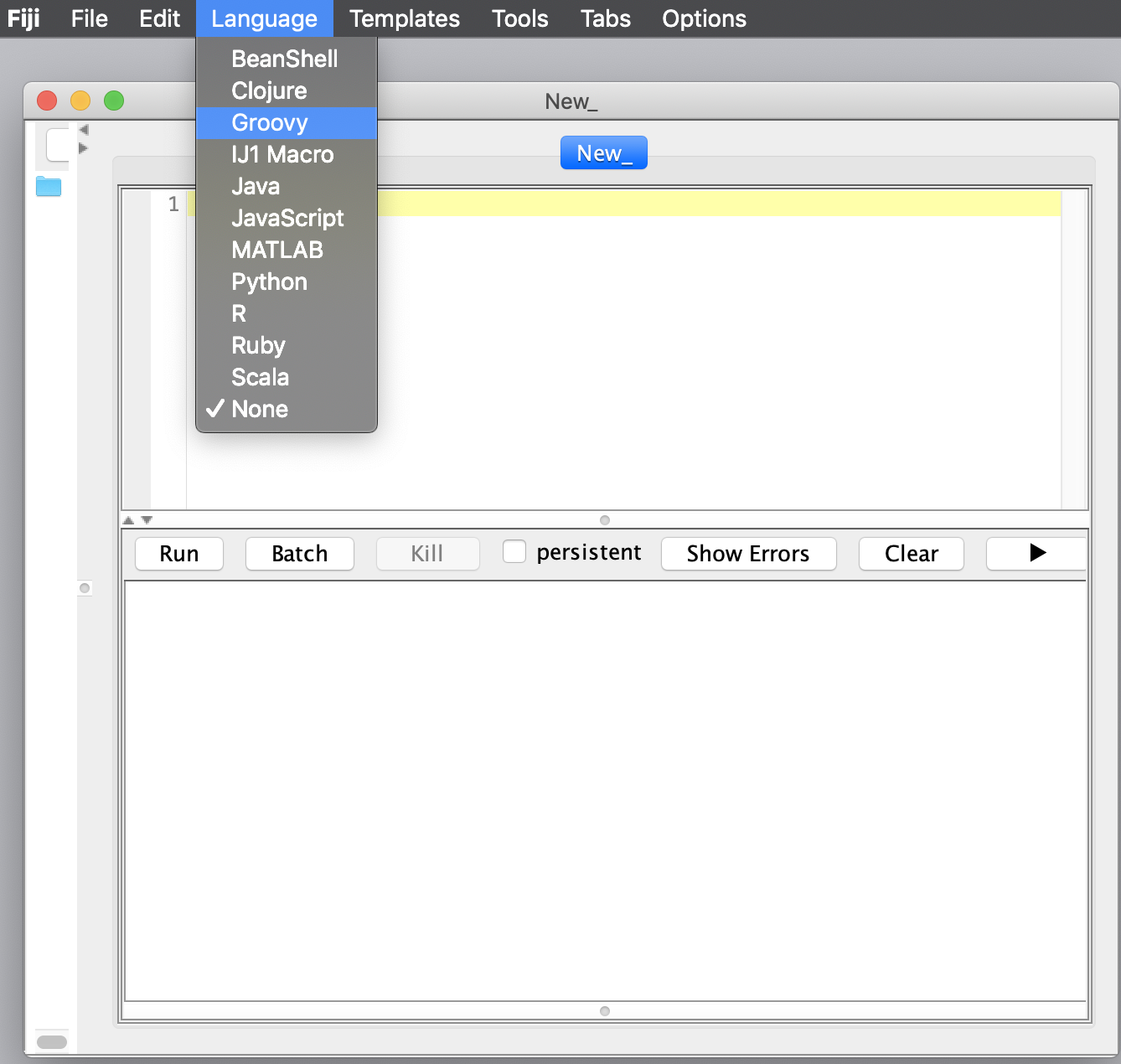
Notice that the script name changes to the .groovy extension after the Groovy language is selected.
2. How to Execute a Script
Next, copy the short example script below and paste it into the scripting window. Run the script by pressing the run button. A dialogue window will pop up in which the archive of interest has to be selected. In this case choose ‘TestVideo_archive.yama’ and press OK. The script will now be run on the archive.
#@ MoleculeArchive archive
sleep(10000)
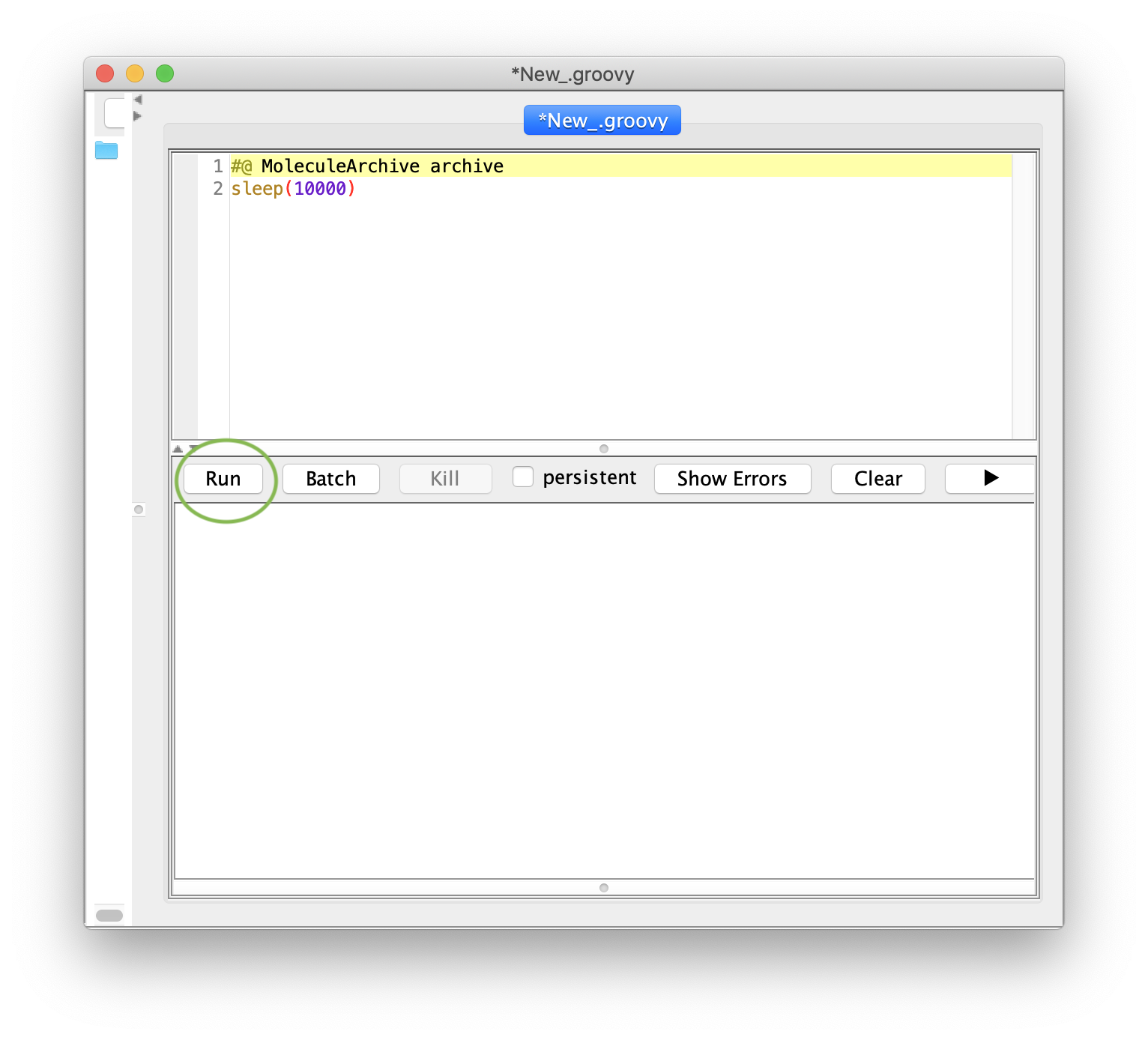
The example script used in this tutorial is a rather illustrative one: it locks the Molecule Archive for 10000 ms (i.e. disables editing functions temporarily) and displays the rotating mars icon to show the user that the archive has been locked. The archive unlocks automatically as soon as the sleep timer has ended.
3. The General Outline of a Script
To learn how write useful scripts, first the general contents of a script are discussed. To make the platform as versatile as possible, Fiji (and therefore Mars) utilises script parameter harvesting. This universal way of denoting input and output parameters allow for an easy interpretation of multiple languages in the software. On top of that, it makes the scripts interpretable in other platforms and script running environments. The syntax uses #@ to denote new parameters and explicitly specifies the parameter type (f.e. string, integer, double, MoleculeArchive) and the name of that parameter.
Script Parameter Syntax
#@ Parametertype Name #define an input parameter
#@ OUTPUT Parametertype Name #define an output parameter
In the example script used before, the parameter ‘archive’ is defined as an input of parameter type ‘MoleculeArchive’. In this way, it is clear to which object the script has to be applied. No output is declared: no output parameters are defined by the script.
4. Access Data from the Molecule Archive with a Script
All functions that can be called on with accompanying documentation can be found in the Mars javadocs. The expected arguments of each function are listed between the brackets of each defined function. Frequently used commands are discussed in this section of the tutorial.
4.1 ‘get’ Functions: get a Parameter from the MoleculeArchive
1 Get the number of molecules in the Molecule Archive
#@ MoleculeArchive archive
archive.getNumberOfMolecules()
2 Get the list of molecule UUIDs
#@ MoleculeArchive archive
println(archive.getMoleculeUIDs())
This prints a comma separated list of the UUIDs in the output window in the lower half of the scripting window. For a neater looking table display of the UUIDs use a MarsTable.
#@ MoleculeArchive archive
#@OUTPUT MarsTable table
import de.mpg.biochem.mars.table.*
import org.scijava.table.*
table = new MarsTable("UIDs")
GenericColumn col = new GenericColumn ("UID")
archive.getMoleculeUIDs().forEach{UID -> col.add(UID)}
table.add(col)
3 Get the list of tags of a certain molecule
#@ MoleculeArchive archive
archive.getTagList('UID')
Replace ‘UID’ by the actual UID of the molecule of interest.
4 Get the value of a parameter. For example the ‘var’ parameter calculated for each molecule entry in the Molecule Archive.
#@ MoleculeArchive archive
archive.get('UID').getParameter('var')
Replace ‘UID’ by the actual UID of the molecule of interest.
5 Get the data table for a molecule entry
#@ MoleculeArchive archive
archive.get('UID').getTable()
Replace ‘UID’ by the actual UID of the molecule of interest.
4.2 ‘has’ Functions: Functions used to check if an Entry has Something
6 Find out if a molecule has a tag
#@ MoleculeArchive archive
archive.moleculeHasTag('UID','tag')
Replace ‘UID’ by the actual UID of the molecule of interest, and ‘tag’ by the actual name of the tag. Returns a boolean indicating whether this molecule has this tag (True) or not (False). Provide only the ‘UUID’ as input to find out if a molecule has any tag.
7 Find out if a molecule has a segments table
#@ MoleculeArchive archive
archive.get('UID').hasSegmentsTable("xcolumn_name","ycolumn_name")
Replace ‘UID’ by the actual UID of the molecule of interest. Returns a boolean indicating whether this molecule has a segments table (True) or not (False).
4.3 General Functions: Save, Store location
8 Save the archive
#@ MoleculeArchive archive
archive.save()
Alternatively, use the saveAs() or saveAsVirtualStore() commands to save the archive with a different name or in virtual storage.
9 Retrieve the archive storage location
#@ MoleculeArchive archive
archive.getStoreLocation()
4.4 ‘set’ Functions: Set Certain Values of the Molecule Archive
10 Define a new parameter in the Molecule Archive
#@ MoleculeArchive archive
archive.get('UID').setParameter('parameter_name',value)
Replace ‘UID’ by the actual UID of the molecule of interest.
11 Set the name the Molecule Archive should be saved to
#@ MoleculeArchive archive
archive.setName('name')
5. Examples for Simple Calculations
5.1 Calculating the total travelled distance in the y direction
In this example the total travelled distance of each molecule in the y direction is calculated and assigned to the parameter ‘dist_y’ declared for all molecule entries. Defining such a parameter might be of use for investigating the heterogeneity in the total distance travelled within the sample and can subsequently be plotted. The following definition is used:
dist_y = max(y) - min(y)
First, the input and output parameters have to be declared: the archive is declared as Molecule Archive type. Since the parameter is assigned directly to the archive as well it does not need to be declared as a separate output. Besides this, the molecule, table, util and scijava table classes are imported.
Next, a loop is defined in which for each UUID the molecule entry is retrieved as well as the table containing coordinates and time points. The ymin and ymax values are calculated by applying the min() and max() functions respectively to the Table on the column named “y”. The distance is calculated as the difference between ymax and ymin and subsequently assigned to the parameter “dist_y”.
#@ MoleculeArchive archive
import de.mpg.biochem.mars.molecule.*
import de.mpg.biochem.mars.table.*
import de.mpg.biochem.mars.util.*
import org.scijava.table.*
archive.getMoleculeUIDs().stream().forEach({UID ->
Molecule molecule = archive.get(UID)
MarsTable table = molecule.getTable()
double ymin = table.min("y")
double ymax = table.max("y")
double dist = ymax - ymin
molecule.setParameter("dist_y",dist)
})
An inspection on the Molecule Archive shows that indeed “dist_y” is added as a parameter to the parameters tab for each molecule.
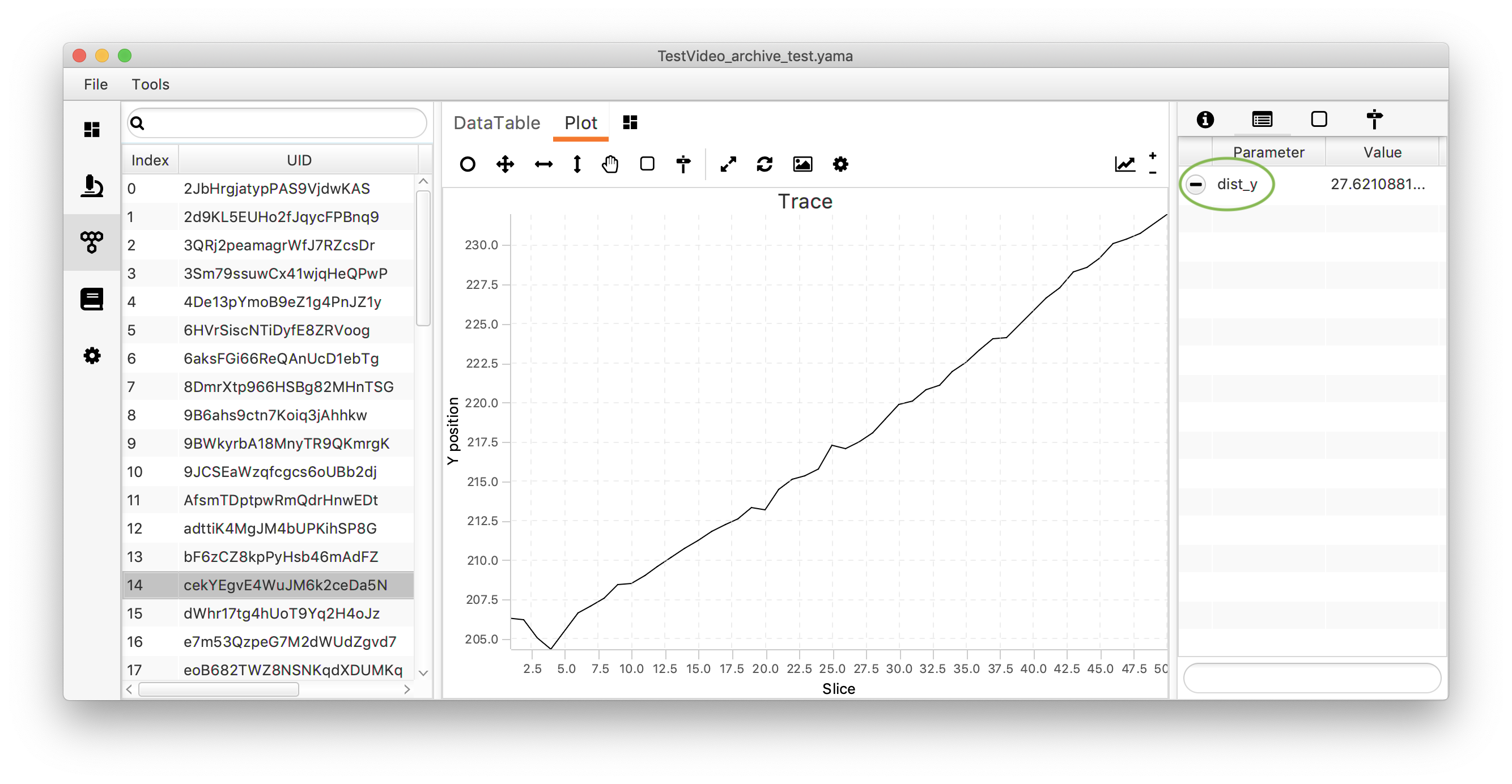
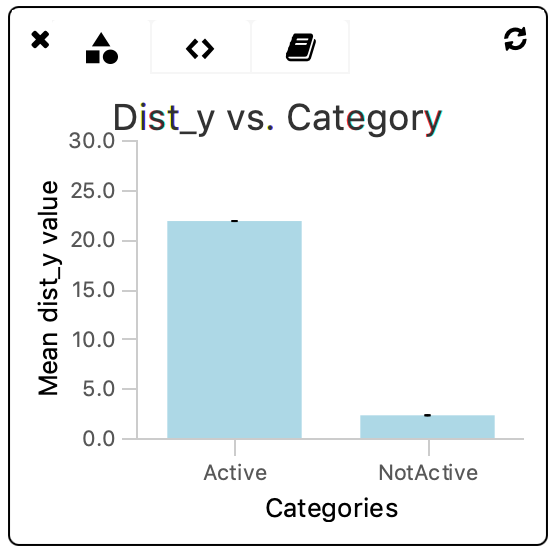
To plot the mean distance travelled in the y direction with respect to tag the category chart widget script in the how to use scriptable widgets tutorial can be adjusted slightly. Pasting the script into the category chart scriptable widget on Rover dashboard yields the chart displayed below. Make sure to select ‘Python’ as the running language for the scriptable widget.
#@ MoleculeArchive archive
#@OUTPUT String xlabel
#@OUTPUT String ylabel
#@OUTPUT String color
#@OUTPUT String title
#@OUTPUT String[] xvalues
#@OUTPUT Double[] yvalues
#@OUTPUT Double ymin
#@OUTPUT Double ymax
color = "#add8e6"
title = "Dist_y vs. Category"
xlabel = "Categories"
ylabel = "Mean dist_y value"
ymin = 0.0
ymax = 30.0
xvalues = ['Active','NotActive'] #Name the categories to be displayed
list1 = []
list2 = []
for UID in archive.getMoleculeUIDs():
if archive.get(UID).hasTag('Active'): #Check if an entry is tagged 'Active'
list1.append(archive.get(UID).getParameter('dist_y'))
else:
list2.append(archive.get(UID).getParameter('dist_y'))
yvalues=[sum(list1)/len(list1),sum(list2)/len(list2)]
The chart shows that there is a clear difference in the travelled distance with respect to the y-axis between molecules tagged ‘Active’ compared to non-tagged molecules. In section 5.2 the variance on the dist_y parameter is calculated.
The Molecule Archive generated in this tutorial can also be found in the tutorial files repository on GitHub.
5.2 Calculate the variance on the travelled distance in the y direction (y_dist)
The sample variance can be calculated according to the following formula:
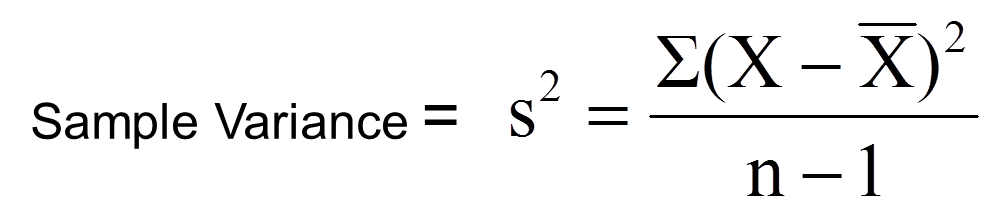
X: data point value (in this case the value of dist_y)
X bar: the mean (mean(dist_y))
n: number of data points
The components of the script to calculate and assign this parameter (dist_y_var) for each molecule in the Molecule Archive is very similar to the previous script. First, the value of dist_y is calculated and assigned to each molecule entry. This step could be omitted in case the script written in section 5.1 already has been run.
#@ MoleculeArchive archive
import de.mpg.biochem.mars.molecule.*
import de.mpg.biochem.mars.table.*
import de.mpg.biochem.mars.util.*
import org.scijava.table.*
def list = []
def list2 = []
archive.getMoleculeUIDs().stream().forEach({UID ->
Molecule molecule = archive.get(UID)
MarsTable table = molecule.getTable()
double ymin = table.min("y")
double ymax = table.max("y")
double dist = ymax - ymin
double[] distlist = []
list.add(dist)
molecule.setParameter("dist_y",dist)
})
double sum = list.sum()
double len = list.size()
double mean = sum/len
archive.getMoleculeUIDs().stream().forEach({UID ->
Molecule molecule = archive.get(UID)
double dist2 = molecule.getParameter("dist_y")
double diff = (dist2 - mean)
double diff2 = diff * diff
list2.add(diff2)
})
double dist_var = list2.sum()/(len-1)
archive.getMetadata(0).setParameter("dist_y_var",dist_var)
As could be expected, the variance is rather high. As shown in section 5.1 the difference in the travelled distance on the y-axis is very different for tagged molecules compared to non-tagged molecules. Therefore, a next step could be to calculate the variance for tagged molecules only, as well as a variance for the non-tagged population. This is further described in the advanced groovy scripting tutorial.
The archive generated in this tutorial can also be found in the tutorial files repository on GitHub (‘TestVideo_archive_disty_var.yama’).
6. Scripting with a virtually stored Molecule Archive
Note that in case an archive is stored virtually each script has to be extended with the line below after a certain operation has been executed to a molecule record (f.e. at the end of the calculation loop in the scripts above).
archive.put(molecule)
In the case of a virtually stored archive, the script retrieves a molecule record to the non-virtual memory, it performs the action as written in the script, returns the record to the virtual storage, and continues. Subsequently, this additional line is required to place the modified record back in virtual storage.