How to copy drift from one archive to another
The Drift Corrector can be used to calculate background drift over the measurement time using a Single Molecule Archive with all immobile molecules tagged as illustrated in the RNA polymerase tracking example. This tutorial will review the steps required to use the Drift Corrector and how to copy the calculated drift to another archive. This allows for the development of drift correction workflows involving other archive types, such as DnaMoleculeArchives, with the molecule tracking done using the tracking options in the BigDataViewer window opened in the Mars Rover.
1. Track molecules
Open your image in Fiji and make sure the channel and time dimensions are correct. Use the Peak Tracker (Plugins>Mars>Image>PeakTracker) to track the positions of many molecules with settings optimized for immobile, surface stuck molecules. When the command is finished a Single Molecule Archive should open containing many complete molecule tracks. This usually included both the desired immobile molecules as well as moving molecules.
Robust calculation of drift is only possible using many position estimates for at each time point. Therefore, make sure the settings used generate molecule tracks with positions for as many time points as possible. Partial tracks are also used during analysis.
2. Calculate variance and tag immobile molecules
Next, use the Variance Calculator for both the X and Y columns separately adding parameters ‘X_Variance’ and ‘Y_Variance’, respectively to all molecule records. Variance distributions can be evaluated using the bar graph widget using the following scripts. These scripts can be copied and pasted into the script panel of the groovy widgets to replace the default script.
For X_Variance:
#@ Context scijavaContext
#@ MoleculeArchive archive
#@OUTPUT String xlabel
#@OUTPUT String ylabel
#@OUTPUT String title
#@OUTPUT Integer bins
#@OUTPUT Double xmin
#@OUTPUT Double xmax
import java.util.stream.Collectors
//Set global outputs
xlabel = "Variance"
ylabel = "Frequency"
title = "X Variance Histogram"
bins = 100
xmin = 0.0
xmax = 5.0
//Series 1 Outputs
#@OUTPUT Double[] series1_values
#@OUTPUT String series1_strokeColor
#@OUTPUT Integer series1_strokeWidth
series1_strokeColor = "red"
series1_strokeWidth = 2
series1_values = archive.molecules().mapToDouble{ mol -> mol.getParameter("X_Variance")}.toArray()
For Y_Variance:
#@ Context scijavaContext
#@ MoleculeArchive archive
#@OUTPUT String xlabel
#@OUTPUT String ylabel
#@OUTPUT String title
#@OUTPUT Integer bins
#@OUTPUT Double xmin
#@OUTPUT Double xmax
import java.util.stream.Collectors
//Set global outputs
xlabel = "Variance"
ylabel = "Frequency"
title = "Y Variance Histogram"
bins = 100
xmin = 0.0
xmax = 30.0
//Series 1 Outputs
#@OUTPUT Double[] series1_values
#@OUTPUT String series1_strokeColor
#@OUTPUT Integer series1_strokeWidth
series1_strokeColor = "blue"
series1_strokeWidth = 2
series1_values = archive.molecules().mapToDouble{ mol -> mol.getParameter("Y_Variance")}.toArray()
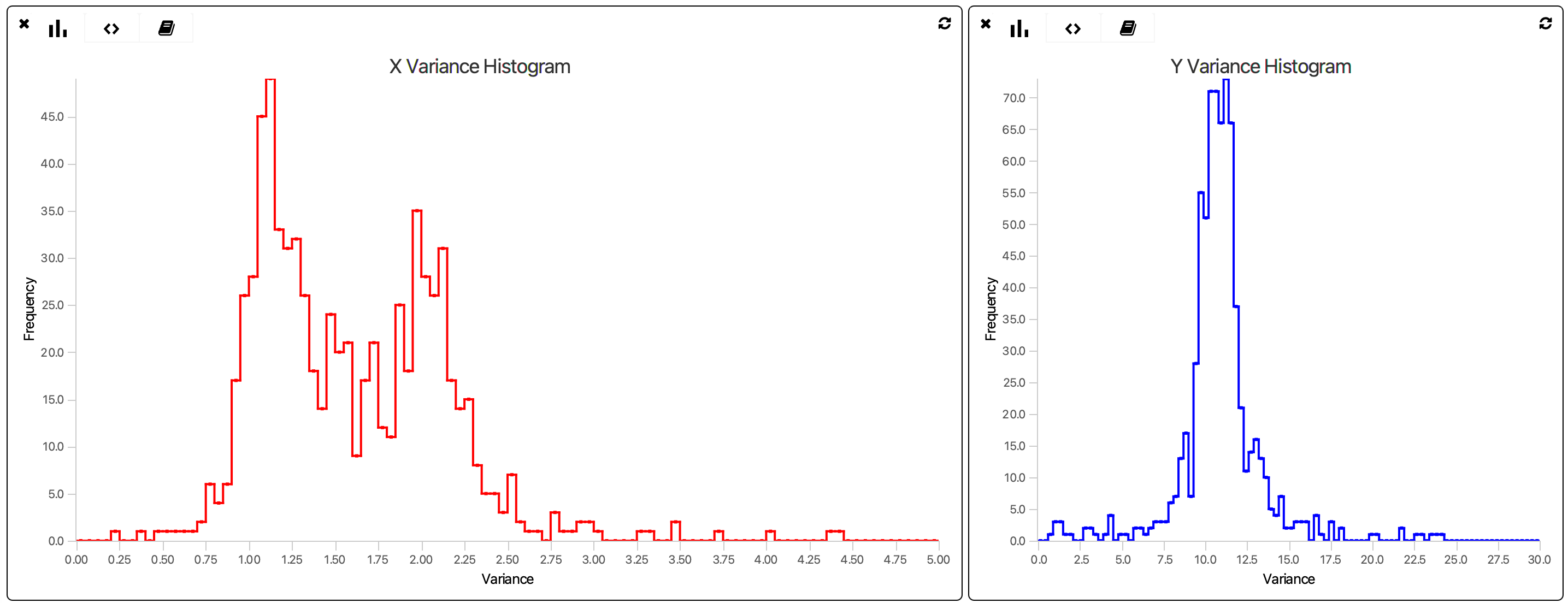
The xmin, xmax, and bins will need to be adjusted depending on the dataset to reveal the distributions. Using visual inspection identify the populations with the lowest variance that correspond to immobile molecules. Use the cutoff values determined in the following script to tag only the immobile molecules. This script should be pasted and run in the Fiji script editor with the language set to Groovy.
#@ MoleculeArchive archive
archive.molecules().filter{ m -> m.getParameter("X_Variance") < 1.5 && m.getParameter("Y_Variance") < 12.5 }.forEach{ m -> m.addTag("background") }
3. Calculate the drift
Now run the Drift Corrector command using the ‘background’ tag to calculate the drift and add the information into the metadata of the archive. You can leave the drift correction checkbox unchecked at the bottom. This is only needed to calculated new drift corrected tracks. After the command is finished, you can check that drift was calculated by going into planes in the metadata and looking at the XDrift and YDrift values.
4. Copy the calculated drift to another archive.
Keep the Single Molecule Archive open that you just created with the drift information. Open the second archive where want to copy the drift information. Run the script below in the Fiji script editor with Groovy chosen for the language. In the dialog that opens select the Single Molecule Archive with the drift information for the option driftArchive and the other archive for toArchive. Running the script should result in the drift information being copied into the metadata of the second archive. This information appears in the values for each plane under XDrift and YDrift. This information can then be used to correct videos for drift in the BDV by turning on drift correction in the BDV settings panel.
#@ MoleculeArchive driftArchive
#@ MoleculeArchive toArchive
driftArchive.getMetadata(0).getImage(0).planes().forEach{ plane ->
int theZ = plane.getZ()
int theC = plane.getC()
int theT = plane.getT()
double driftX = plane.getXDrift()
double driftY = plane.getYDrift()
toArchive.metadata().forEach{metadata ->
metadata.getImage(0).getPlane(theZ, theC, theT).setXDrift(driftX)
metadata.getImage(0).getPlane(theZ, theC, theT).setYDrift(driftY)
}
}